
le Syndrome de Joubert est un trouble d'origine génétique qui se caractérise par une diminution du tonus musculaire, des problèmes de coordination, des mouvements oculaires anormaux, des schémas respiratoires modifiés et une déficience intellectuelle (Joubert Syndrome Foundation, 2016).
Toutes ces altérations sont dues à une transmission génétique autosomique qui entraînera des anomalies cérébrales importantes, une réduction du vermis cérébelleux, ainsi que des anomalies dans la structure du tronc cérébral (National Institute of Neurological Disorders and Stroke, 2016).
De plus, le syndrome de Joubert fait partie d'un groupe de troubles appelés ciliopathies qui impliquent un dysfonctionnement d'une partie des cellules appelées cils. Fondation du syndrome de Joubert, 2016).
La description initiale de cette pathologie a été faite par Marie Joubert et al.en 1968, dans laquelle quatre cas ont été décrits. Chez les patients, il y avait absence partielle ou totale du vermis cérébelleux, syndrome d'ampnée-hypernée épisodique néonatal, mouvements oculaires anormaux, ataxie et retard mental (Angemi et Zucotti, 2012).
De plus, ce syndrome était également associé à différentes altérations multiorganiques, telles que la fibrose hépatique, la polydactylie, la néphronoptysie ou la dystrophie rétinienne (Angemi et Zucotti, 2012).
En termes de traitement, il n'existe actuellement aucun remède pour le syndrome de Joubert. Les interventions thérapeutiques visent le contrôle et le soutien des symptômes, la stimulation physique et intellectuelle des enfants et l'ergothérapie (National Institute of Neurological Disorders and Stroke, 2016).
Index des articles
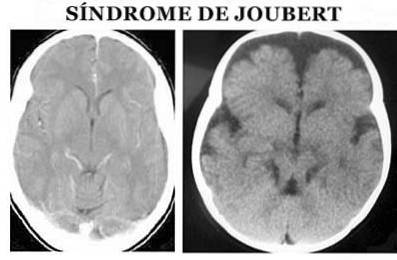
Le syndrome de Joubert (JS) est un type de pathologie d'origine génétique qui se caractérise par une malformation congénitale dans les zones du tronc cérébral et une agénésie (absence partielle ou complète) ou une hypoplasie (développement incomplet) du vermis cérébelleux, qui peut provoquer (Ophatnet , 2009).
Plus précisément, au niveau anatomique, il est caractérisé par le signe dit molaire du mésencéphale: agénésie ou hypoplasie du vermis cérébelleux, rétrécissement des pédoncules cérébelleux supérieurs avec épaississement, allongement et absence de décussation et fosse interpédonculaire profonde (Angemi et Zuccoti, 2012).
C'est un trouble qui peut affecter de nombreuses zones et organes du corps, de sorte que les signes et les symptômes varient considérablement selon les personnes touchées (U.S. National Library of Medicine, 2011).
La plupart des personnes atteintes souffrent d'un affaiblissement du tonus musculaire (hypotonie) et de difficultés de coordination motrice (ataxie). Les autres traits caractéristiques sont: des épisodes de respiration altérée, un nystagmus (mouvement involontaire et arythmique des yeux), un retard du développement moteur et des difficultés intellectuelles variables (U.S. National Library of Medicine, 2011).
La prévalence du syndrome de Joubert a été estimée entre 1/80 000 et 1/100 000 000 naissances vivantes. Dans le monde, plus de 200 cas cliniques ont été enregistrés (Angemi et Zuccoti, 2012).
De nombreux spécialistes considèrent que ces chiffres sont sous-estimés, car le syndrome de Joubert a un large éventail d'affections et est largement sous-diagnostiqué (U.S. National Library of Medicine, 2011).
La plupart des symptômes cliniques du syndrome de Joubert sont plus qu'évidents dans l'enfance, de nombreux enfants atteints présentent des retards moteurs importants (National Organization for Rare Disease, 2011).
Les caractéristiques les plus courantes de l'évolution clinique sont: un manque de contrôle musculaire (ataxie), une modification des schémas respiratoires (hypercapnie), l'apnée du sommeil, des mouvements oculaires anormaux (nystagmus) et un faible tonus musculaire (National Organization for Rare Disease, 2011).
En revanche, certaines des altérations pouvant être associées au syndrome de Joubert comprennent: une altération du développement de la rétine, des anomalies de l'iris, un strabisme, des altérations rénales et / ou hépatiques, une protrusion des membranes qui recouvrent le cerveau, entre autres ( Organisation nationale pour les maladies rares, 2011).
Toutes les altérations dérivées de ce syndrome sont englobées dans plusieurs domaines: altérations neurologiques, oculaires, rénales et musculo-squelettiques (Bracanti et al., 2010).
Les altérations neurologiques les plus caractéristiques du syndrome de Joubert sont Bracanti et al., 2010): hypotonie, ataxie, retard généralisé du développement, altérations intellectuelles, altération des schémas respiratoires et mouvements oculaires anormaux.
Sur le plan physique, la rétine est l'un des organes touchés par le syndrome de Joubert. Les altérations de cet organe apparaissent sous forme de dystrophie rétinienne, due à une dégénérescence progressive des cellules responsables de la photo réception.
Cliniquement, les altérations oculaires peuvent aller de la cécité rétinienne congénitale à la dégénérescence rétinienne progressive.
D'autre part, il est également possible d'observer la présence du colobome. Cette altération oculaire est une anomalie congénitale qui affecte l'iris oculaire et se présente sous la forme d'un trou ou d'une fente.
Les pathologies liées à la fonction rénale touchent plus de 25% des personnes atteintes du syndrome de Joubert.
Dans de nombreux cas, les anomalies rénales peuvent rester asymptomatiques pendant plusieurs années ou commencer à se manifester par des signes non spécifiques, jusqu'à ce qu'elles se présentent comme une insuffisance rénale aiguë ou chronique..
Dès les premières descriptions de cette pathologie, un constat clinique fréquent est la polydactialie (une maladie génétique qui augmente le nombre de doigts ou d'orteils).
De plus, il est également fréquent d'observer des anomalies orofaciales ou structurelles au niveau de la colonne vertébrale.
Des études expérimentales ont classé le syndrome de Joubert comme un trouble autosomique récessif (National Organization for Rare Disease, 2011).
Un trouble génétique autosomique récessif signifie que deux copies d'un gène anormal doivent être présentes pour que le trait ou la maladie se présente (National Institutes of Health, 2014).
Par conséquent, une altération génétique récessive se produit lorsqu'une personne hérite du même gène anormal pour le même trait de chaque parent. Si un individu ne reçoit qu'une seule copie du gène lié à la maladie, il sera porteur mais ne présentera pas de symptômes (National Organization for Rare Disease, 2011).
De plus, au moins dix gènes ont été identifiés comme l'une des causes possibles du syndrome de Joubert (National Organization for Rare Disease, 2011).
Une mutation du gène AHI1 est responsable de cette pathologie chez environ 11% des familles touchées. Chez les personnes atteintes de cette altération génétique, les altérations de la vision sont courantes en raison du développement d'une dystrophie rétinienne (National Organization for Rare Disease, 2011).
La mutation du gène nphp1 est à l'origine d'environ 1 à 2% des cas de syndrome de Joubert. Chez les personnes présentant cette altération génétique, les altérations rénales sont courantes (National Organization for Rare Disease, 2011).
En revanche, une mutation du gène CEP290 est à l'origine de 4 à 10% des cas de syndrome de Joubert (National Organization for Rare Disease, 2011).
De plus, des mutations dans les gènes TME67, JBTS1, JBTS2, JBTS7, JBTS8 et JBTS9 sont également liées au développement du syndrome de Joubert (National Organization for Rare Disease, 2011).
Le diagnostic du syndrome de Joubert est posé sur la base des symptômes physiques. Il est nécessaire d'effectuer à la fois un examen physique détaillé, ainsi que l'utilisation de différents tests de diagnostic, en particulier des images de résonance magnétique (Ophatnet, 2009).
De plus, les tests de génétique moléculaire sont également souvent utilisés pour identifier des altérations génétiques qui ont été démontrées dans 40% des cas de syndrome de Joubert (National Organization for Rare Disease, 2011).
D'autre part, il est également possible de faire un diagnostic prénatal de cette pathologie par échographie fœtale et analyse moléculaire, notamment dans les familles ayant des antécédents génétiques de syndrome de Joubert (Ophatnet, 2009).
Lorsque les caractéristiques les plus caractéristiques du syndrome de Joubert surviennent en association avec une ou plusieurs pathologies physiques supplémentaires, un diagnostic de syndrome de Joubert et de troubles associés (JSRD) peut être posé (U.S. National Library of Medicine, 2011).
Par conséquent, en fonction du type de pathologie associée associée à la présence du syndrome de Joubert, nous pouvons en trouver des sous-types. Cependant, le système de classification du syndrome de Joubert est encore en phase d'évolution en raison de la découverte de contributions génétiques et de la meilleure connaissance des corrélations phénotypiques..
On peut donc trouver (Bracanti et al., 2010):
Le traitement utilisé dans le syndrome de Joubert est symptomatique et supporte les pathologies sous-jacentes. En plus des interventions pharmacologiques, il est courant d'utiliser une stimulation physique et cognitive précoce (National Institute of Neurological Disorders and Stoke, 2016).
Lorsque les altérations respiratoires sont importantes, en particulier dans les phases initiales de la vie, il est nécessaire de surveiller la fonction respiratoire (National Institute of Neurological Disorders and Stoke, 2016).
En revanche, l'identification et le contrôle de la dégénérescence oculaire, des complications rénales et du reste des complications liées au syndrome de Joubert, doivent être réalisés le plus tôt possible pour ajuster les mesures thérapeutiques (National Institute of Neurological Disorders and Stoke, 2016 ).


Personne n'a encore commenté ce post.