La Maladie de Canavan C'est une maladie génétique rare qui survient parce que les cellules nerveuses du cerveau sont endommagées et incapables de communiquer entre elles. Cette maladie est présente dans n'importe quelle société et groupe ethnique, bien qu'elle soit beaucoup plus fréquente dans la population juive ashkénaze et ses descendants, où 1 personne sur 6 400 à 13 000 est touchée. La prévalence mondiale est inconnue.
Cette maladie fait partie du groupe des leucodystrophies. Cette catégorie comprend tous les troubles génétiques dans lesquels la gaine de myéline qui entoure les axones des neurones est endommagée et, par conséquent, il n'y a pas de bonne communication entre les neurones.
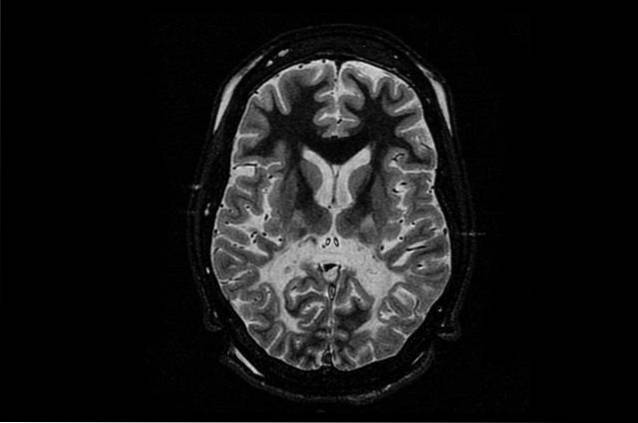
La forme la plus courante et, en même temps, la plus grave de cette maladie est néonatale ou infantile. Cette forme de maladie de Canavan affecte les nouveau-nés ou dans leurs premières années de vie..
Les enfants qui souffrent de cette maladie ne présentent aucun problème pendant les premiers mois de la vie, mais ceux-ci commencent à fleurir entre 3 et 5 mois. Les principaux symptômes sont dus au déficit de développement, où les enfants ont des problèmes moteurs qui les empêchent de se retourner, de tourner la tête ou de s'asseoir sans aucun soutien..
D'autres symptômes courants sont la faiblesse musculaire (hypotonie), le développement anormal de la tête (macrocéphalie) et l'irritabilité. Dans une moindre mesure, ils peuvent également avoir des difficultés à manger, des convulsions et des problèmes de sommeil..
Une autre forme moins courante est la maladie de Canavan qui commence au milieu de l'enfance ou à l'adolescence. Les enfants et adolescents atteints de cette maladie ont des problèmes de développement du langage et de motricité, mais ces problèmes sont souvent si bénins qu'ils ne sont pas identifiés comme des symptômes de la maladie de Canavan..
L'espérance de vie des personnes atteintes de la maladie de Canavan est très hétérogène, variant notamment selon l'heure d'apparition de la maladie.
Les enfants qui souffrent de la forme néonatale ou infantile ne vivent généralement que quelques années, bien que certains atteignent l'adolescence et très peu jusqu'à l'âge adulte. Alors que ceux qui souffrent de la forme juvénile ont une espérance de vie normale.
Index des articles
Il existe deux formes bien différenciées de la maladie de Canavan: celle d'apparition néonatale ou infantile et celle d'apparition au milieu de l'enfance ou de l'adolescence..
Les symptômes de la maladie de Canavan néonatale ou infantile sont très graves, généralement visibles avant l'âge de 3 à 50 mois, et comprennent une macrocéphalie, une perte de contrôle moteur de la tête et des déficits de développement. Les déficits de développement deviennent plus apparents à mesure que l'enfant grandit.
Les symptômes les plus graves sont ceux liés aux problèmes moteurs, car les enfants sont incapables de s'asseoir ou de se lever sans soutien, marcher ou parler. Lorsqu'ils vieillissent, l'hypotonie peut entraîner une spasticité.
Bien qu'ils aient tous ces problèmes moteurs, ils peuvent apprendre à interagir socialement, sourire, pointer des objets ...
Certains enfants souffrent également d'atrophie optique, ce qui entraîne des problèmes visuels, bien qu'ils puissent toujours identifier les objets visuellement.
À mesure que les symptômes s'aggravent, ils s'aggravent, provoquant des troubles du sommeil, des convulsions et des problèmes d'alimentation. L'enfant devient totalement dépendant, a besoin d'aide pour effectuer n'importe quelle tâche.
L'espérance de vie de ces enfants est assez courte, la plupart meurent en quelques années, même si certains vivent jusqu'à l'adolescence ou à l'âge adulte.
La maladie de Canavan apparaissant au milieu de l'enfance ou de l'adolescence est plus bénigne que la précédente. Les symptômes comprennent certaines difficultés de développement verbal et moteur.
Bien qu'ils soient généralement si bénins qu'ils ne sont pas identifiés comme des symptômes de la maladie de Canavan, cette maladie est généralement diagnostiquée après une analyse d'urine, car l'un des marqueurs est la concentration élevée d'acide N-acétylaspartique (NAA dans l'urine)..
Cette maladie est causée par une mutation dans un gène appelé ASPA. Ce gène est celui qui contrôle l'enzyme aspartoacylase, responsable de la dégradation des molécules NAA..
La mutation du gène ASPA amène l'aspartoacylase à réduire son efficacité, elle ne dégradera donc pas suffisamment les molécules de NAA et il y aura une concentration élevée de cette substance. Plus cette mutation se produit tôt, plus elle a de mauvais effets.
Bien que le fonctionnement des molécules NAA ne soit pas très bien compris, il semble qu'elles soient impliquées dans le transport des molécules d'eau à travers les neurones et, l'excès de cette substance, empêche la formation de nouvelle myéline et détruit l'existant. Cela fait que les connexions entre les neurones ne fonctionnent pas correctement et que le cerveau est incapable de se développer normalement..
De plus, cette maladie peut être héréditaire de manière autosomique récessive. Ainsi, si chaque membre du couple est porteur de la variante pathogène du gène ASPA et décide d'avoir un enfant, il est susceptible de:
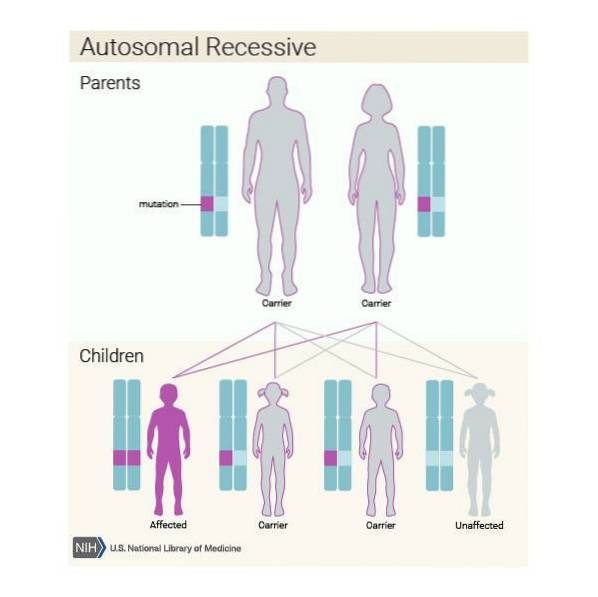
Il est très important que les individus appartenant à la population à risque, en l'occurrence les descendants de juifs ashkénazes, subissent une analyse génétique pour vérifier s'ils sont porteurs du gène ASPA avant d'avoir un enfant..
Le traitement dépend de la forme de la maladie et des symptômes que chaque individu présente..
Il n'y a actuellement aucun remède pour la maladie de Canavan, donc les thérapies disponibles se concentrent sur l'amélioration de la qualité de vie du patient en soutenant, nourrissant et hydratant, et en prévenant et traitant les infections.
Il est recommandé que les enfants reçoivent un traitement physiothérapeutique pour améliorer leur posture et leur motricité, pour éviter et traiter les contractures et les problèmes musculaires, tels que les escarres. Ils peuvent également participer à des programmes thérapeutiques et éducatifs pour améliorer leurs compétences en communication..
Le traitement médicamenteux comprend des antiépileptiques (AED) si l'enfant a des convulsions, l'acétazolamide (nom de marque Diamox®) pour réduire la pression intracrânienne et les injections de toxine botulique (Botox®) pour traiter la spasticité si présente.
Il est nécessaire de faire un suivi tous les 6 mois pour vérifier dans quel état se trouve l'enfant et comment se déroule son développement.
Les personnes qui souffrent de cette forme de la maladie éprouvent des symptômes beaucoup plus légers, de sorte qu'elles n'ont généralement besoin que de thérapies pour améliorer leur langue ou de programmes éducatifs spéciaux. Ils n'ont besoin d'aucun médicament.
Une surveillance annuelle de l'état de l'enfant est recommandée.
L'efficacité d'autres thérapies est actuellement étudiée sur des modèles humains et animaux..
L'efficacité d'une transplantation génétique dans le cerveau d'enfants atteints de la maladie de Canavan est à l'étude, en utilisant un vecteur non viral.
Les premiers résultats montrent que ce type de transplantation est bien toléré par les enfants et provoque des changements biochimiques, radiologiques et métaboliques, mais il n'est pas utile pour guérir la maladie, donc des tests sont toujours en cours (Leone et al 2000, Janson et al .à 2002).
McPhee et coll. (2006) mènent une étude dans laquelle le gène ASPA sain est transplanté à divers endroits du corps des enfants, en utilisant l'AAV2 comme vecteur. Dans l'un des tests auxquels 10 enfants volontaires ont participé. Dans 3 d'entre eux, la greffe a fonctionné et neutralisé leurs anticorps, mais aucun des enfants ne s'est amélioré.
Le citrate de lithium peut réduire le niveau de concentration de NAA dans le cerveau, c'est pourquoi Assadi et al. (2010) ont décidé de mener une expérience dans laquelle ils ont administré du citrate de lithium à 6 personnes atteintes de la maladie de Canavan pendant 60 jours..
Des niveaux de concentration de NAA dans les noyaux gris centraux et la substance blanche du lobe frontal ont été trouvés, bien qu'aucune amélioration clinique n'ait été trouvée.
Le manque d'enzymes aspartoacylase provoque de faibles niveaux d'acétate dans le cerveau, alors Mahavarao et son équipe (2009) ont décidé de donner du triacétate de glycérol à deux patients atteints de la maladie de Canaval pour augmenter leurs niveaux d'acétate et voir si cela augmentait également les niveaux d'aspartoacylase..
Le composé a été bien toléré par les patients, bien qu'aucune amélioration clinique n'ait été trouvée. Ils mènent actuellement des tests administrant une quantité plus élevée de triacétate de glycérol.
L'une des façons de créer des modèles animaux qui représentent une maladie est de créer des animaux Assommer. Ces animaux, généralement des souris, sont génétiquement modifiés pour supprimer ou modifier le gène altéré par la maladie. Dans ce cas, le gène modifié est le gène ASPA..
Des modèles animaux sont utilisés pour mieux comprendre la maladie, étudier son corrélat biologique et vérifier l'efficacité des nouveaux traitements.
Matalon et coll. (2003) ont utilisé des souris Assommer pour tester l'efficacité d'une thérapie génique avec AAV2 comme vecteur. Ils ont constaté qu'il y avait eu des améliorations dans les gaines de myéline, mais seulement dans certaines parties, pas dans tout le cerveau..
L'équipe de Surendran, en collaboration avec la Genzyme Corporation (2004), a testé un traitement de greffe de cellules souches. Ils ont constaté que de nouveaux oligodendrocytes avaient été produits, mais pas suffisamment pour restaurer toutes les gaines de myéline..
Une autre équipe a testé une thérapie qui consistait à remplacer les enzymes asparthoacyclases défectueuses par de nouvelles qui étaient injectées dans le péritoine des souris. Assommer.
Les résultats à court terme ont montré que les enzymes étaient capables de franchir la barrière hémato-encéphalique (atteignant leur objectif) et étaient capables de réduire considérablement les niveaux de NAA dans le cerveau. Bien que ces résultats soient prometteurs, une étude longitudinale est nécessaire pour vérifier les effets à long terme (Zano et al., 2011).
Les premiers signes qui alertent les médecins que quelque chose ne va pas sont les signes physiques, en particulier l'hypotonie et la macrocéphalie..
Normalement, si ces signes sont observés, une étude de neuro-imagerie est généralement réalisée chez l'enfant pour vérifier les signes de leucodystrophie, comme une densité plus faible de substance blanche. Il est à noter que ce test est moins efficace chez les enfants atteints de la maladie de Canavan qui commence au milieu de l'enfance ou à l'adolescence..
Une fois qu'il a été prouvé que l'enfant souffre d'une leucodystrophie, des tests plus spécifiques sont effectués pour exclure d'autres maladies, notamment:
La dernière étape pour confirmer la maladie serait d'effectuer une étude génétique comme suit:



Personne n'a encore commenté ce post.