La phakomatose est un groupe de troubles neurocutanés d'origine génétique, rares dans la population générale. Au niveau clinique, ils se caractérisent par le développement d'une atteinte organique multisystémique avec des lésions cutanées ou tumorales, dans différentes zones de la peau, des organes ou du système nerveux..
De plus, son évolution clinique non spécifique rend son diagnostic précoce difficile, de sorte que ses conséquences médicales et psychologiques détériorent considérablement la qualité de vie de la personne touchée et de ses proches..
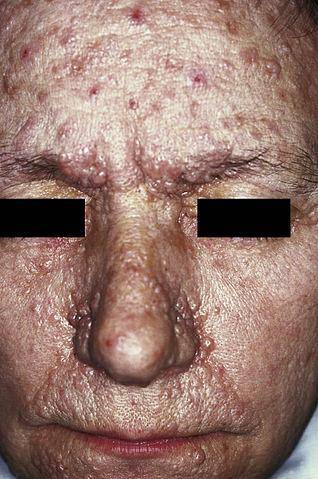
Bien qu'il existe un grand nombre de maladies neurocutanées, les plus courantes comprennent la fibromatose de type I et de type II, la maladie de Bourneville, le syndrome de Sturge-Weber et la maladie de Von Hippel-Lindau..
D'autre part, malgré le fait que toutes ces pathologies soient congénitales, de multiples approches thérapeutiques de nature dermatologique ont été conçues pour tenter d'améliorer les signes et symptômes caractéristiques de ces troubles et, par conséquent, le pronostic médical des personnes atteintes..
Index des articles
Le terme phakomatose vient de l'expression d'origine grecque Phakos dont le sens se réfère à <
Les pathologies neurocutanées sont fondamentalement caractérisées par l'existence d'une association significative entre une affectation ou un trouble neurologique et les manifestations dermatologiques.
Ainsi, le terme pathologie neurocutanée est utilisé de manière généralisée pour englober différentes maladies présentes chez la personne atteinte congénitale et qui, en outre, peuvent être présentes tout au long de la vie avec le développement de lésions cutanées et de tumeurs dans différentes zones, système nerveux, système cardiovasculaire, système rénal, système cutané, système ophtalmique, etc..
De cette manière, le terme phakomatose a été introduit en 1917 par Brouwer et plus tard par van der Hoeve en 1923, cependant, les descriptions initiales ne faisaient référence qu'à certaines pathologies incluses dans ce groupe. Actuellement, plus de 40.
Au niveau clinique, la phakomatose est décrite comme une maladie qui présente des altérations cutanées et des malformations bénignes / malignes dans différents systèmes: neurologique, oculaire, cutané et viscéral..
Concernant les zones touchées, divers auteurs soulignent que celles d'origine ectodermique sont les plus touchées, c'est-à-dire la peau et le système nerveux, bien qu'elles puissent également affecter d'autres systèmes ou dispositifs, comme l'œil..
Les syndromes et pathologies d'origine neurocutanée sont des maladies rares dans la population générale, bien qu'il n'y ait pas de données spécifiques à un niveau général sur toutes ces maladies.
Ainsi, l'épidémiologie de ces troubles varie en fonction du type de maladie, en particulier, la neurofibromatose est l'une des plus courantes, avec une prévalence relative d'un cas pour 300 000 naissances.
Les maladies neurocutanées sont caractérisées par le développement de lésions cutanées. Plus précisément, la phakomatose se distingue de beaucoup d'autres par la présence d'hamartomes..
Les hamartomes sont un type de malformation ou de tumeur bénigne qui peut se développer dans différents organes tels que le cerveau, le cœur, les yeux, la peau ou les poumons..
Cependant, la phakomatose peut être associée à un grand nombre de conditions médicales qui varieront, fondamentalement, en fonction de la maladie ou de la pathologie spécifique subie par la personne affectée..
Actuellement, un grand nombre de troubles neurocutanés ont été identifiés au niveau clinique et génétique, mais il en existe certains avec une prévalence plus élevée dans la population générale: neurofibromatose de type I et de type II, maladie de Bourneville, maladie de Von Hippel-Lindau et Sturge-Weber syndrome.
Il existe différentes formes cliniques de neurofibromatose. Cependant, à l'heure actuelle, les plus fréquentes sont la neurofibromatose de type I, également appelée maladie de Von Reclinghausen, et la neurofibromatose de type II, suivie de la shwannomatose rachidienne..
Au niveau étiologique, toutes ces manifestations médicales de la neurofibromatose ont une origine génétique et se produisent avec la formation de tumeurs dans les zones nerveuses, en particulier le système nerveux central et périphérique..
Les formations tumorales, généralement non cancéreuses ou bénignes, ont tendance à se développer et à se développer presque partout dans le système nerveux, comme le cerveau, la moelle épinière ou les nerfs périphériques.
Ainsi, les algues de complications médicales secondaires à la neurofibromatose comprennent des anomalies de croissance, le développement de convulsions, l'apparition de tumeurs cérébrales, des pathologies osseuses, la surdité et / ou la cécité, ou le développement de problèmes d'apprentissage importants, entre autres..
De plus, cette pathologie est présente dès la naissance. Cependant, la manifestation significative de son tableau clinique peut être retardée jusqu'à la fin de la petite enfance, au début de l'adolescence ou à l'âge adulte..
En revanche, le diagnostic de ce type de pathologie comprend généralement, outre l'examen physique et neurologique, différents tests de neuroimagerie et analyses génétiques..
De plus, à l'heure actuelle, il n'y a pas de remède pour la neurofibromatose, cependant, il existe des approches thérapeutiques spécialisées dans le contrôle de l'affectation dermatologique, elles peuvent inclure des traitements pharmacologiques et chirurgicaux pour arrêter ou éliminer les formations tumorales..
La neurofibromatose de type I (NF1), également connue sous le nom de maladie de von Recklinghausen, se manifeste principalement par la présence de taches brun clair, communément appelées «café au lait», d'éphélides (taches de rousseur) et de neurofibromes (lésions nerveuses des cellules de Schwann et des neurites).
Il a une origine génétique autosomique dominante, spécifiquement due à une mutation sur le chromosome 17, à l'emplacement 17q11.2. Ainsi, le gène impliqué dans
le développement de la neurofibromatose de type I, a un rôle de premier plan dans la modulation de la croissance et de la différenciation cellulaires et, en outre, peut fonctionner comme un suppresseur de tumeur.
Concernant l'épidémiologie de cette pathologie, elle présente une prévalence approximative d'un cas pour 2.500.3000 naissances.
Le diagnostic de neurofibromatose de type I est généralement posé sur la base des critères cliniques consensuels du National Institute of Health (1987), cependant, il nécessite un suivi continu pour éviter des complications médicales secondaires..
Normalement, les excroissances tumorales sont traitées avec des médicaments, pour empêcher leur développement exponentiel ou par ablation chirurgicale.
La neurofibromatose de type II (NF2), se manifeste principalement par le développement de schwannomes, c'est-à-dire de formations tumorales dérivées de cellules de Shcwaan qui seront chargées de couvrir les prolongations nerveuses.
Les schwannomes ou neuriomes touchent généralement en particulier le nerf auditif et optique et dans une moindre mesure les zones cutanées.
La neurofibromatose de type II a une origine génétique autosomique dominante, en particulier en raison de la présence d'une mutation sur le chromosome 22, à l'emplacement 22q11.22.
Le gène impliqué dans le développement de cette pathologie est responsable du codage d'un composant protéique ayant un rôle de premier plan dans la suppression tumorale, de sorte que son activité déficiente produit une augmentation anormale de la prolifération cellulaire.
Concernant l'épidémiologie de cette pathologie, elle est moins fréquente que le type 1, présentant une prévalence approximative d'un cas pour 50000 naissances.
Le diagnostic de neurofibromatose de type II est similaire au type précédent et est généralement posé sur la base des critères cliniques consensuels de l'Institut national de la santé. Cependant, il comprend généralement des tests de laboratoire complémentaires, tels que la neuroimagerie..
Normalement, les excroissances tumorales sont traitées avec des médicaments, cependant, dans les cas où cela est possible, une ablation chirurgicale est utilisée.
La maladie de Bourneville est l'un des termes utilisés pour désigner la sclérose tubéreuse, une maladie génétique caractérisée par la présence d'hamartomes.
Au niveau clinique, elle peut donner lieu à une affectation multisystémique caractérisée par une affectation cutanée (angiomes faciaux, fibromes de l'ongle, plaques fibreuses, taches hypochromiques, etc.), une atteinte rénale (angiomyolipomes rénaux ou kystes rénaux), une atteinte cardiaque (rhabdomyomes cardiaques) , affectation neurologique (tubercules corticaux, nodules gliaux sous-épendymaires, atrocytomes, convulsions, déficience intellectuelle, anomalies comportementales et motrices), entre autres.
Comme les maladies décrites ci-dessus, l'origine de la sclérose tubéreuse est génétique. Plus précisément, cela est dû à la présence de mutations dans les gènes TSC1 et TSC2..
En revanche, le diagnostic de sclérose tubéreuse est posé sur la base des critères cliniques proposés lors d'une conférence médicale en 1998. Cependant, l'étude génétique est également considérée comme pertinente pour sa confirmation.
En ce qui concerne le traitement de la sclérose tubéreuse, bien qu'il n'y ait pas de remède, différentes approches pharmacologiques et chirurgicales sont généralement utilisées, principalement pour le contrôle des croissances tumorales et des complications médicales secondaires telles que les manifestations neurologiques..
La maladie de Von Hippel-Lindau, également appelée angiomatose rétino-cérébelleuse, se manifeste principalement par la présence et le développement de malformations vasculaires, de kystes et / ou de tumeurs, généralement bénignes..
Il a une origine génétique autosomique dominante, spécifiquement due à une mutation sur le chromosome 3, à l'emplacement 3p-25-26. En outre, il présente une incidence estimée à un cas pour 40 000 naissances..
Plus précisément, la maladie de Von Hippel-Lindau affecte principalement le système nerveux central (SNC) et la rétine, à travers la formation d'hémangiomes.
Les hémangiomes sont des malformations vasculaires caractérisées par la présence d'amas de capillaires sanguins dilatés. Ils apparaissent généralement dans les zones du cerveau et de la colonne vertébrale, bien qu'ils soient également fréquents dans les rétines ou sur la peau.
Le diagnostic de cette pathologie, en plus de l'examen physique et neurologique, nécessite une étude ophtalmologique détaillée, ainsi que l'analyse de différents tests de neuroimagerie, pour confirmer la présence de lésions nerveuses..
Concernant le traitement de la maladie de Von Hippel-Lindau, l'intervention de base est la chirurgie pour éliminer les malformations vasculaires. Cependant, il nécessite une surveillance continue pour éviter les complications secondaires..
De plus, il a une espérance de vie réduite, autour de 50 ans, principalement en raison du développement de carcinomes rénaux (formations néoplasiques de cellules cancéreuses dans les tubules rénaux).
Le syndrome de Sturge-Weber, également connu sous le nom d'angiomatose encéphalo-trijumeau, se manifeste principalement par la présence d'hémangiomes.
Un hémangiome est un type de néoplasme ou de formation de tumeur caractérisé par la présence d'un nombre anormalement élevé de vaisseaux sanguins dans la peau ou d'autres organes internes..
Plus précisément, au niveau clinique, le syndrome de Sturge-Weber est caractérisé par le développement d'hémangiomes faciaux, d'hémangiomes intracrâniens et d'hémangiomes choridiques, conjonctivaux, épiscéraux et glaucome..
Il a une origine génétique, spécifiquement due à une mutation sur le chromosome 9, à l'emplacement 9q21, dans le gène GNQ. Cette composante génétique a un rôle de premier plan dans le contrôle des facteurs de croissance, des peptides vasoactifs et des neurotransmetteurs (Orhphanet, 2014).
Le diagnostic du syndrome de Sturge-Weber repose sur des soupçons cliniques et différents tests de laboratoire, tels que la tomographie informatisée ou l'imagerie par résonance magnétique..
D'autre part, en termes de traitement, la thérapie au laser est capable de réduire la progression de cette pathologie et, en outre, dans de nombreux cas, d'éliminer complètement les hémangiomes..

Personne n'a encore commenté ce post.