Les prions ce sont des protéines sans génome ou des acides nucléiques qui agissent comme des agents infectieux. Le terme «prion» signifie particule infectieuse protéique (de l'anglais Proteinaceous Infectious Particles), et a été inventé par le neurologue et lauréat du prix Nobel, Stanley B. Prusiner.
En 1982, Prusiner et ses collègues ont identifié une particule de protéine infectieuse, tout en étudiant les causes des maladies de Creutzfeldt-Jakob (chez l'homme) et de l'encéphalopathie spongiforme bovine..
Ces agents infectieux rares se trouvent dans la membrane des cellules normales, uniquement sous forme de protéines mal repliées et / ou avec une structure tridimensionnelle anormale. Ces protéines sont responsables de multiples maladies dégénératives et d'une mortalité très élevée qui affectent les tissus neuraux et la structure du cerveau..
Ils sont également appelés maladies à prions. Le kuru, la maladie de Gerstmann-Sträussler-Scheinker, le syndrome de Creutzfeldt-Jakob et l'insomnie familiale fatale sont parmi les plus importants qui affectent les humains..
Index des articles
Les prions sont des structures protéiques présentes dans les membranes cellulaires. Ces protéines ont une forme ou une conformation modifiée [PrP (Sc)].
Quant à sa multiplication, elle se fait par la conversion des formes, comme dans le cas de la tremblante du mouton. Dans cette maladie, les prions recrutent PrP (C) (protéines prions de conformation inchangée) pour stimuler la conversion en isoforme PrP (Sc)..
Cela génère une réaction en chaîne qui propage le matériel infectieux et permet donc l'irrigation de la maladie. On ne sait toujours pas comment ce processus de conversion se produit.
Ces protéines inhabituelles capables de se propager, ne possèdent pas d'acides nucléiques. La preuve en est qu'ils sont résistants aux rayons X et aux rayons ultraviolets. Ces agents décomposent facilement les acides nucléiques.
Les protéines prions, dont les prions (PrP) sont composés, se trouvent dans tout le corps, non seulement chez l'homme mais chez d'autres vertébrés sains. Ces protéines sont généralement résistantes aux protéases (enzymes qui catalysent les protéines).
On sait très peu de choses sur l'utilité des protéines prions PrP (C), la forme normale de la protéine non infectieuse dans le corps humain..
Cependant, certains chercheurs ont réussi à montrer que, chez la souris, ces protéines activent la réparation de la myéline dans les cellules du système nerveux périphérique. Il a également été démontré que l'absence de ceux-ci provoquait la démyélinisation de ces cellules nerveuses..
La connaissance que l'on a de la structure des prions réside principalement dans les investigations menées sur la bactérie Escherichia coli.
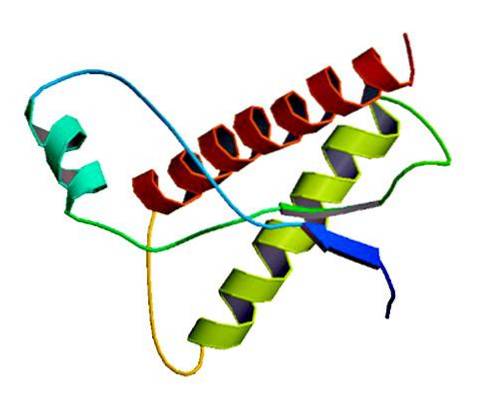
Des études ont montré que les polypeptides de chaîne PrP (C) (normal) et PrP (Sc) (infectieux) sont identiques dans la composition en acides aminés, mais diffèrent dans leur conformation 3D et leur repliement..
Ces prions non infectieux ont 209 acides aminés chez l'homme. Ils ont une liaison disulfure. Sa structure est en hélice alpha, ce qui signifie qu'elle possède des acides aminés en forme de spirale (hélices alpha) et quelques brins plats d'acides aminés (feuillets bêta)..
Cette protéine ne peut pas être séparée par centrifugation, ce qui implique qu'elle n'est pas sédimentable. Il est facilement digéré par la sérine protéase à large spectre appelée protéinase K.
C'est une protéine infectieuse qui transforme la PrP (C) en isoformes infectieuses de la PrP (Sc) et avec une configuration ou une forme anormale.
On sait très peu de choses sur sa structure 3D, mais on sait qu'elle a peu de formes hélicoïdales et plus de brins plats ou de feuilles bêta. Le passage à l'isoforme est ce que l'on appelle l'événement pivot des maladies à prions.
Les protéines prions cellulaires [Prp (C)] sont situées à la surface cellulaire d'une grande variété d'organes et de tissus. On en sait très peu sur les fonctions physiologiques des prions dans le corps. Même ainsi, les expériences réalisées sur la souris indiquent des fonctions possibles, telles que:
Il a été démontré que la PrP (C) agit avec les récepteurs du glutamate (ionotropes et métabotropiques). PrP (C) participe en tant que récepteur pour les oligomères synaptotoxiques du peptide de surface cellulaire Aβ.
Chez les souris de la famille Murinae, il a été découvert que les protéines prions PrP (C) sont exprimées en quelques jours après l'implantation, dans le développement embryonnaire..
Cela indique qu'ils jouent un rôle lors du développement de ces petits mammifères. Rôle qui selon les chercheurs est lié à la régulation de la neuritogenèse (production d'axones et de dendrites de neurones).
Ils agissent également sur la croissance axonale. Ces protéines prions sont même impliquées dans le développement du circuit cérébelleux. Pour cette raison, on pense que l'absence de ces prions PrP (C) entraîne un retard dans le développement moteur des rongeurs..
Dans des études sur la surexpression de la PrP (C) par orientation génique, il a été constaté que l'absence de ces prions entraîne des problèmes d'approvisionnement en sang vers certaines parties du cerveau (ischémie cérébrale aiguë).
Cela signifie que les protéines prions fonctionnent comme des neuroprotecteurs. De plus, il a été démontré que la surexpression de PrP (C) peut réduire ou améliorer les blessures causées par l'ischémie..
Le rôle physiologique de Prp (C) dans le maintien de la myéline périphérique a été récemment découvert.
Au cours d'une étude en laboratoire, il a été découvert qu'en l'absence de protéine prion, les souris de laboratoire développaient des déficiences dans les nerfs qui transportent les informations du cerveau et de la moelle épinière, dans ce qu'on appelle une neuropathie périphérique..
Certaines protéines sont similaires aux prions, et celles-ci sont situées dans d'autres parties du corps que le cerveau.
Les fonctions de telles protéines sont d'initier, de réguler et / ou de contrôler la mort cellulaire, lorsque l'organisme est attaqué (par des virons par exemple), empêchant ainsi la propagation du pathogène..
Cette fonction particulière de ces protéines amène les chercheurs à réfléchir à l'importance possible des prions non infectieux dans la lutte contre les agents pathogènes..
Une étude menée au Stowers Institute, dans le Missouri, aux États-Unis, a montré que les prions PrP peuvent jouer un rôle dans le maintien de la mémoire à long terme..
L'étude a révélé que certaines protéines prions peuvent être contrôlées pour travailler au maintien des fonctions physiologiques de la mémoire à long terme..
Une enquête sur les protéines prions exprimées dans les cellules souches des tissus sanguins, a révélé que toutes ces cellules souches (hématopoïétiques) expriment des protéines prions dans leur membrane cellulaire. Pour ce que l'on pense qu'ils participent au processus complexe et très important de renouvellement cellulaire.
Les pathologies d'origine prionique sont reconnues comme des troubles dégénératifs progressifs du cerveau. Ils peuvent attaquer les bovins, les cerfs, les caribous, les moutons et même les humains.
Ces maladies sont causées par une altération de la structure des protéines PrP (C) et dont les fonctions spécifiques sont encore aujourd'hui incertaines. Les pathologies à prions peuvent survenir sans cause connue. Ils peuvent avoir une origine génétique héréditaire et peuvent également être transmis de manière infectieuse-contagieuse.
Les prions provoquent des maladies familiales, sporadiques et contagieuses. Les maladies familiales à prions sont celles qui sont héréditaires. Les pathologies sporadiques sont les plus courantes et surviennent sans causes connues..
Les maladies contagieuses sont considérées comme rares, elles se transmettent de personne à personne, d'animal à animal, de personne à animal et vice versa. Les causes sont multiples et vont de la consommation de viande contaminée, au cannibalisme, aux transfusions, à la manipulation de matériel chirurgical contaminé.
Les maladies à prions les plus courantes sont:
Considérée comme la maladie à prion la plus répandue chez l'homme, c'est une maladie cosmopolite, c'est-à-dire qu'elle a une distribution mondiale. Il peut être héréditaire (familial), sporadique ou infectieux.
Les patients présentent des symptômes tels que démence, secousses ou mouvements involontaires soudains et déficiences du système nerveux central.
Selon le traitement et la forme de la maladie, le décès peut survenir entre 4 mois et 2 ans après l'acquisition de la maladie. Le diagnostic est difficile à faire, il est généralement fait post morten, pendant l'autopsie.
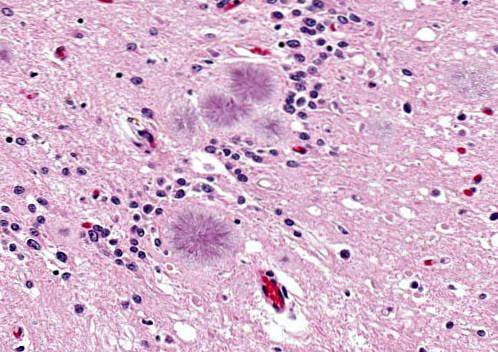
C'est une maladie causée par des prions dans un processus cérébral infectieux héréditaire ou autosomique dominant. La maladie se manifeste chez les personnes âgées de 40 à 60 ans.
Ces personnes manifestent des problèmes d'articulation des mots (dysarthrie), des saccades ou des mouvements involontaires soudains, l'agressivité étant fréquente.
Ils présentent une dégénérescence cérébelleuse accompagnée d'une démarche instable. Il est également possible d'observer une hyporéflexie, une surdité, une paralysie du regard, une démence, entre autres symptômes. L'espérance de vie est d'environ 5 ans ou un peu plus.
C'est une maladie très rare, au point que sa fréquence d'apparition est de 2 à 3 cas pour 100 millions d'habitants. La pathologie est similaire à la maladie de Gerstmann-Sträussler-Scheinker.
Les manifestations cliniques de la protéine indiquent une faible résistance aux protéases, certaines sont plus et d'autres moins sensibles à ces enzymes.
Les symptômes que présentent les patients sont: des problèmes d'élocution et de troubles cognitifs, une perte de neurones dans la zone où le cerveau contrôle les mouvements et effectue la coordination musculaire.
La maladie est fréquente chez les patients âgés (70 ans) et la durée de vie estimée une fois infectée est d'environ 20 mois.
C'est une maladie héréditaire ou familiale, elle peut également survenir de manière sporadique. La maladie est connue pour être due à une mutation héréditaire ou autosomique dominante.
Les patients présentent des symptômes tels que des problèmes cumulatifs de sommeil et de maintien du sommeil, de démence, de troubles cognitifs, voire des problèmes d'hypertension, de tachycardie, d'hyperhidrose et autres..
L'âge qu'il affecte est assez large, allant de 23 à 73 ans, mais l'âge moyen est de 40 ans. La durée de vie une fois infectée est d'un peu plus de 6 ans.
Cette maladie à prions n'a été détectée que chez les habitants de Papouasie-Nouvelle-Guinée. C'est une maladie liée au cannibalisme et à la tradition culturelle du rite du deuil des morts, où ces personnes mangent du cerveau ou de la chair humaine.
Les personnes porteuses de la maladie ont généralement des mouvements incontrôlables et involontaires dans différentes parties du corps.
Ils présentent des tremblements, une perte de contrôle des mouvements et une perte de coordination musculaire. L'espérance de vie des personnes infectées est de deux ans.
L'encéphalopathie spongiforme bovine fait partie des pathologies produites par les prions chez les animaux. Cette maladie a fait des ravages en Europe, dans la santé publique, celle des animaux et dans l'économie des pays touchés.
D'autres maladies chez les animaux comprennent la tremblante, l'encéphalopathie transmissible du vison, la maladie débilitante chronique (chez le cerf) et l'encéphalopathie spongiforme féline..
Ces maladies, comme celles qui se présentent chez l'homme, n'ont pas de traitement efficace, la prévention est donc essentielle, en particulier après des infections chez l'homme survenues à la suite de la consommation de viande de vaches infectées..
À ce jour, il n'existe aucun remède connu pour les maladies à prions. Le traitement est symptomatique. Les patients sont invités à planifier des soins palliatifs et des tests génétiques et des conseils pour les membres de la famille sont recommandés.
Une grande variété de médicaments ont été testés chez des patients atteints de maladies à prion, tels que les antiviraux, les antitumeurs, les médicaments pour des maladies comme la maladie de Parkinson, les traitements pour l'immunosuppression, les antibiotiques, les antifongiques, voire les antidépresseurs..
Cependant, il n'existe actuellement aucune preuve indiquant que certains d'entre eux réduisent les symptômes ou améliorent la survie des patients..
Les prions sont résistants à une variété de changements physiques et chimiques. Cependant, différentes techniques sont utilisées pour éviter la contamination des patients par des instruments chirurgicaux contaminés..
L'une des techniques les plus utilisées consiste à stériliser le matériel dans un autoclave à 132 ° C pendant une heure puis à immerger les instruments dans de l'hydroxyde de sodium pendant au moins une heure de plus..
D'autre part, l'Organisation mondiale de la santé (OMS) a mis au point des mesures pour prévenir la propagation des maladies à prions. Cet organisme établit des normes pour la manipulation de tissus interdits ou potentiellement à risque tels que: les yeux, le cerveau, l'intestin, les amygdales et la moelle épinière.


Personne n'a encore commenté ce post.