le Syndrome de Cornelia de Lange est une pathologie d'origine génétique qui se caractérise par la présence d'un retard cognitif important accompagné de divers traits physiques malformatifs.
Au niveau clinique, trois évolutions cliniques différentielles sont observées: sévère, modérée et légère. Les signes et symptômes sont généralement constitués par une configuration faciale atypique, des malformations musculo-squelettiques et un retard du développement cognitif et psychomoteur. De plus, d'autres types d'anomalies liées à des malformations cardiaques, pulmonaires et / ou digestives peuvent être distinguées..
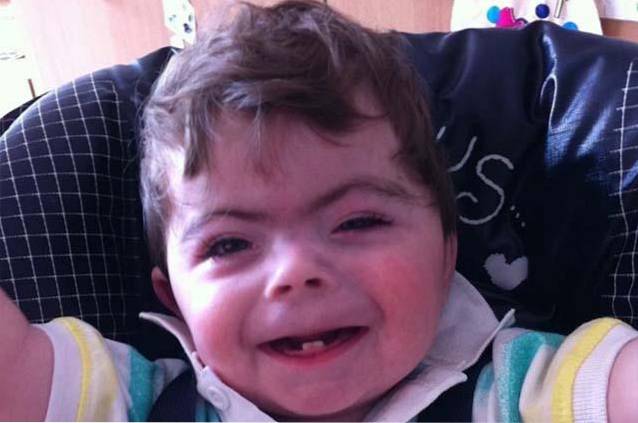
Concernant l'origine du syndrome de Cornelia de Lange, son étiologie a été liée à la présence de mutations spécifiques dans les gènes SMC3, SMC1A, NIPBL, entre autres. Le diagnostic est fondamentalement clinique, fait sur la base de caractéristiques physiques et cognitives. Cependant, il est généralement accompagné d'un test génétique de confirmation.
Le traitement est orienté vers la détection et le traitement des complications médicales. La médecine, l'orthophonie, l'intervention neuropsychologique et l'éducation spéciale sont essentielles.
Index des articles
Ce syndrome a été initialement décrit par le Dr Cornelia de Lange en 1933. Ses recherches étaient basées sur l'étude de deux patients âgés de 6 et 17 mois. Son tableau clinique était caractérisé par un retard sévère de la croissance physique et du développement intellectuel associé à divers traits malformatifs..
Compte tenu de la similitude des deux cas, le premier rapport clinique sur cette pathologie supposait l'existence d'une cause étiologique commune et publique.
Auparavant, Brachmann (1916) avait réussi à publier des données d'autopsie d'un patient en âge d'enfant présentant certaines caractéristiques compatibles avec le syndrome de Cornelia de Lange..
À l'heure actuelle, le tableau clinique de ce syndrome a été classé en trois phénotypes différentiels: sévère, modéré et léger..
Le syndrome de Cornelia de Lange est une maladie génétique rare de nature congénitale, c'est-à-dire que ses caractéristiques cliniques sont évidentes dès la naissance. Elle est définie comme une maladie multisystémique avec des symptômes associés à un retard du développement physique et cognitif, des malformations cranio-faciales ou des malformations musculo-squelettiques..
Bien que l'évolution clinique et la gravité de ce syndrome puissent varier considérablement parmi les personnes touchées, il s'agit d'une maladie avec un taux de mortalité élevé..
Les personnes atteintes du syndrome de Cornelia de Lange se caractérisent par une configuration faciale atypique ou caractéristique et un retard de croissance / développement pré et postnatal..
Le plus courant est l'apparition de problèmes d'apprentissage, un retard d'acquisition du langage ou de la démarche et des anomalies comportementales.
Le syndrome de Cornelia de Lange est une maladie rare dans la population générale, elle est généralement classée dans les maladies rares. Les données épidémiologiques ne sont pas exactement connues. Son incidence a été estimée à un cas pour 10 000 à 30 000 naissances..
À ce jour, nous pouvons trouver plus de 400 cas différents de syndrome de Cornelia de Lange décrits dans la littérature médicale et expérimentale..
C'est une pathologie qui peut toucher les deux sexes en nombre égal. Certains auteurs comme Gutiérrez Fernández et Pacheco Cumani (2016) suggèrent une légère prédominance envers les femmes, avec un ratio de 1,3 / 1.
En ce qui concerne le reste des facteurs sociodémographiques, la recherche actuelle n'a pas identifié de prévalence différentielle associée à des pays ou à des groupes ethniques et / ou raciaux spécifiques..
Une bonne partie des cas diagnostiqués sont sporadiques, bien que plusieurs familles touchées aient été identifiées avec un modèle clair d'hérédité dominante..
Les signes et symptômes du syndrome de Cornelia de Lange sont caractérisés par leur large schéma d'implication.
Cette maladie est définie par la présence de traits caractéristiques du visage, des malformations musculo-squelettiques des membres supérieurs et inférieurs, un retard de croissance généralisé pré et postnatal, ainsi que le développement d'autres anomalies physiques..
Ensuite, nous décrirons certaines des caractéristiques cliniques les plus fréquentes du syndrome de Cornelia de Lange:
Chez plus de 90% des personnes atteintes du syndrome de Cornelia Lange, il est possible d'identifier un retard du développement physique ou une hypogrowth globale. La croissance est généralement affectée à la fois avant et après la naissance.
Les caractéristiques les plus courantes chez les nouveau-nés sont:
Ces conditions durent généralement jusqu'à l'âge adulte. On y distingue une croissance située en dessous de celle attendue pour le sexe et l'âge biologique de la personne affectée.
Parallèlement à ce type d'altérations, certaines anomalies liées à l'alimentation peuvent être identifiées. La difficulté à avaler ou à mâcher de la nourriture est courante pendant les premiers stades de la vie.
La combinaison d'altérations crâniennes et faciales entraîne le développement d'un phénotype facial caractéristique chez les personnes atteintes du syndrome de Cornelia de Lange..
Certaines des anomalies les plus courantes comprennent:
Le retard du développement cognitif et psychomoteur constitue l'un des principaux résultats cliniques du syndrome de Cornelia Lange. Une lente acquisition de compétences liées à l'activité motrice ou mentale est généralement identifiée.
Les jalons les plus touchés sont l'acquisition de l'assise, le sourire affectif, le babillage, le mouvement indépendant, l'émission des premiers mots, la compréhension et les ordres, l'alimentation, la déambulation ou la toilette indépendante.
Dans beaucoup de personnes touchées, un QI moyen associé à une déficience intellectuelle légère ou modérée peut être identifié.
Le comportement des personnes atteintes du syndrome de Cornelia de Lange présente généralement des traits distinctifs:
Le syndrome de Cornelia de Lange est également associé au développement de diverses complications médicales.
Les causes les plus fréquentes de décès ou d'aggravation de l'état de santé des personnes touchées sont liées à:
La variabilité des signes et symptômes du syndrome de Cornelia de Lange a permis une classification de son évolution clinique:
C'est généralement le plus grave. Les altérations et anomalies sont caractérisées par la présence de sous-bois physique, des malformations musculo-squelettiques, des traits du visage anormaux, une limitation de la mobilité articulaire, un retard cognitif et d'autres complications médicales (auditives, oculaires, digestives, réno-urologiques, cardiaques et génitales)..
Dans ce sous-type, les altérations physiques ne sont généralement pas très évidentes, en particulier dans les extrémités. Les personnes touchées ne présentent généralement pas de déficits intellectuels graves. Le plus courant est que le diagnostic est posé au-delà du stade néonatal.
Son évolution clinique est fondamentalement caractérisée par une variabilité clinique. Des caractéristiques faciales sont présentes dans la plupart des cas, mais l'expression du reste des anomalies est variable..
L'origine du syndrome de Cornelia Lange est associée à la présence d'anomalies génétiques. Dans les cas examinés, il a été possible d'identifier des mutations spécifiques dans 5 gènes différents: NIPBL, SMC1A, HDAC8, RAD21 et SMC3.
L'altération la plus courante est liée au gène NIPBL, identifié chez plus de la moitié des personnes touchées. Le reste des anomalies génétiques est moins fréquent.
Tous ces gènes jouent un rôle prépondérant dans la production de protéines liées au complexe de la cohésine, responsables de la régulation de la structure et de l'organisation chromosomiques, de la stabilisation de l'information génétique dans les cellules et de la réparation de l'ADN..
En outre, ils remplissent également plusieurs fonctions fondamentales dans le développement prénatal des extrémités, du visage et d'autres régions et systèmes corporels..
Le diagnostic du syndrome de Cornelia de Lange est clinique. Actuellement, aucun test de laboratoire n'indique définitivement sa présence. Dans le domaine médical, le plus courant est d'utiliser les critères diagnostiques proposés par Kline et al..
Celles-ci concernent l'identification des anomalies cranio-faciales, de la croissance et du développement, des extrémités, des altérations neurosensorielles et cutanées, des troubles du comportement, etc..
De plus, il est important de réaliser une analyse génétique moléculaire pour identifier la présence de mutations associées au syndrome de Cornelia de Lange..
Bien qu'il n'y ait pas de remède pour le syndrome de Cornelia de Lange, son approche thérapeutique implique la conception d'un suivi médical continu ainsi que le traitement des complications..
Les auteurs Gil, Ribate et Ramos (2010) soulignent certaines des approches les plus utilisées.



Personne n'a encore commenté ce post.