
le Syndrome de DiGeorge est une pathologie d'origine génétique qui se manifeste par le développement de malformations liées à la structure du cœur, du visage, du thymus et des glandes parathyroïdes.
Au niveau clinique, ils produiront une grande variété de complications médicales, parmi lesquelles les déficiences immunitaires, l'hypocalcémie, les pathologies cardiaques et les troubles psychiatriques..

En ce qui concerne l'origine étiologique, il est associé à une altération génétique du chromosome 22. De ce fait, il est également appelé syndrome de délétion 22q11.2..
Le diagnostic repose sur l'identification des signes cliniques cardinaux par l'examen physique et divers tests de laboratoire: examen analytique et immunologique, échographie abdominale, échocardiogrammes et étude génétique, fondamentalement basée sur l'hybridation fluorescente in situ (FISH)..
Enfin, le traitement de cette pathologie se concentre sur la correction des malformations organiques et le contrôle des complications médicales. Ainsi, la thérapie des lymphocytes T, les suppléments de calcium, la chirurgie corrective, etc. sont généralement utilisés..
Index des articles
Cette pathologie a été initialement décrite par le pédiatre américain Angelo M. DiGeorge en 1965. Dans son rapport clinique, DiGeroge a décrit une pathologie congénitale définie par le développement déficient ou l'absence de la glande parathyroïde et du thymus.
Par la suite, Chapelle en 1918, décrit spécifiquement les malformations congénitales dérivées de cette pathologie. Ainsi, le syndrome de DiGeorge a été qualifié de deuxième cause de malformations cardiaques congénitales après le syndrome de Down.
Enfin, cette pathologie a été caractérisée cliniquement par la triade classique de l'immunodéficience, de l'endocrinopathie avec hypocalcémie et des maladies cardiaques..
De plus, dans de nombreux cas, la grande hétérogénéité symptomatique des délétions localisées sur le chromosome 22 implique la différenciation de trois types différents de pathologies au niveau clinique:
- Syndrome de DiGeorge
- Syndrome vélocardiofacial
- Syndrome Cardiofacial
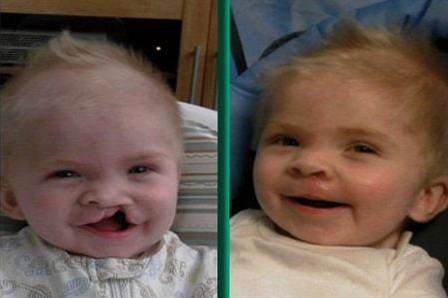
Le syndrome de DiGeorge, également connu sous le nom de syndrome de délétion 22q11.2, est une maladie causée par un défaut génétique qui entraîne le développement de diverses malformations corporelles et organiques..
En ce sens, ce syndrome dérive fondamentalement de processus de développement défectueux pendant la phase prénatale ou gestationnelle, localisés principalement au cours de la 3e et 8e semaine de gestation..
Plus précisément, vers la 5ème semaine de gestation, les structures embryonnaires entament un processus de formation et de développement de différentes structures et organes (Vera de Pedro et al., 2007).
Ainsi, un groupe de certaines cellules donnera lieu au développement du visage, de diverses parties du cerveau, du thymus, du cœur, de l'aorte et des glandes parathyroïdes..
Ce "champ de cellules" est généralement situé autour de la zone ou de la zone située derrière le cou de l'embryon en gestation. De cette manière, pour que le reste des structures commence à se former et à se différencier, il est essentiel que ces cellules se déplacent vers les différentes zones spécifiques de chaque structure..
Dans cette phase de développement, les bourses pharyngées, les arcades et les fissures, le thymus et les glandes parathyroïdes sont formés et plus tard, une partie des structures crâniennes et faciales ou diverses parties du tissu conjonctif.
De cette manière, les anomalies génétiques typiques du syndrome de DiGeroge donnent lieu à une altération systématique de ce processus de formation prénatale, entraînant de graves échecs de développement..
En conséquence, les zones les plus touchées sont généralement:
- Coeur: cette structure constitue l'un des organes vitaux de notre survie. Il fait partie du système circulatoire et sa fonction essentielle est de pomper le sang vers le reste du corps.
- Configuration faciale: la formation de la structure faciale dépend de la formation correcte du crâne, des globes oculaires, du système buccal, des oreilles, etc..
- Escroquer: cette structure joue un rôle fondamental au sein du système immunitaire, puisqu'elle est responsable de la maturation des lymphocytes ou des cellules T.
- Glandes parathyroïdes: ils sont constitués d'un ensemble de glandes endocrines qui jouent un rôle dans la régulation du calcium, entre autres facteurs.
Ainsi, les zones les plus touchées dans le syndrome de DiGeorge sont liées au défaut de formation embryonnaire dans les zones associées au cou et aux régions adjacentes..
Le syndrome de DiGeroge a une prévalence estimée à 1 cas pour 4000 personnes dans la population générale.
Cependant, de nombreuses études épidémiologiques indiquent une prévalence plus élevée principalement en raison de l'hétérogénéité de son évolution clinique et de la difficulté à établir un diagnostic précoce..
De plus, aux États-Unis et dans le monde, le syndrome de DiGeorge est considéré comme l'une des causes les plus courantes de malformations cardiaques congénitales et de malformations faciales..
En revanche, en termes de caractéristiques épidémiologiques de nature sociodémographique, une prévalence de 1 cas pour 6000 personnes d'origine caucasienne, asiatique et afro-descendante a été identifiée, alors que dans le cas des Hispaniques, la prévalence s'élève à un cas pour tous les 3800 individus.
Dans le cas des signes et symptômes les plus fréquents du syndrome de DiGeorge, il faut souligner qu'il présente une évolution clinique avec une expressivité variable.
Dans ce cas, chez certains patients, les complications médicales présentent un état grave, ce qui peut entraîner une mort prématurée. Dans d'autres cas, les caractéristiques présentent généralement un compromis minimal pour la survie et la fonctionnalité de la personne affectée..
Par conséquent, toutes les personnes touchées par le syndrome de Di George ne présenteront pas la même affectation, mais elles incluent généralement une ou plusieurs altérations associées..
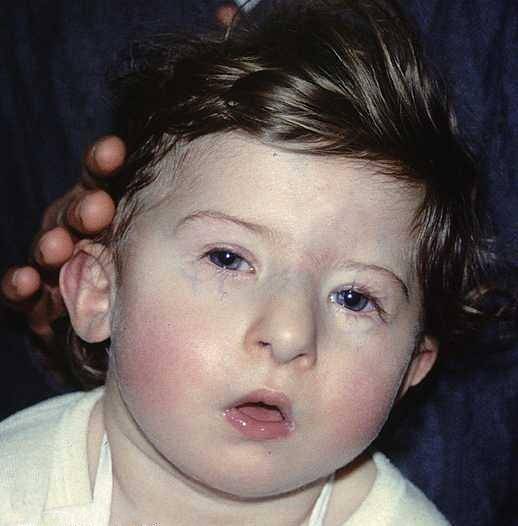
Les altérations liées à la configuration faciale constituent l'une des caractéristiques visuelles les plus frappantes du syndrome de DiGeorge, généralement définies par:
- Microcéphalie: la tête se développe avec une dimension plus petite ou plus petite que prévu pour le niveau de développement et l'âge chronologique de la personne affectée. De plus, une structure nasale tubulaire se développe généralement accompagnée de joues plates ou mal accentuées.
- Hyploplasie mandibulaire et rétrognathie: la structure de la mâchoire n'est pas complètement développée. Ainsi, dans de nombreux cas, il a une taille réduite ou une position modifiée, située plus en arrière que d'habitude..
- Altération oculaire: généralement les yeux ont tendance à être inclus vers le plan inférieur, de plus, une microphtalmie (sous-développement d'un des globes oculaires), des cataractes (opacité du cristallin oculaire) ou une cyanose (coloration bleue) peuvent apparaître autour des yeux.
- Altération du pavillon: il est possible d'identifier une asymétrie dans la configuration des oreilles. Ils ont généralement une faible implantation avec la présence de malformations dans les lobes et d'autres zones externes du pavillon.
- Malformations orales: la configuration de la bouche présente généralement un aspect cambré vers le plan supérieur, caractérisé par la présence d'un sillon nasogénien long et accentué et d'une fente palatine.
Les anomalies cardiaques comprennent souvent une grande variété de défauts. Cependant, les zones les plus touchées sont liées à l'aorte et aux structures cardiaques associées:
- Défauts septaux: la paroi ou la structure qui sépare les cavités cardiaques responsables du pompage du sang peut être formée de manière incomplète ou défectueuse.
- Malformation de l'arc aortique: Diverses anomalies peuvent également être décrites dans le segment aortique situé entre les voies ascendantes et descendantes.
- Tétralogie de Fallot: cette pathologie fait référence à la présence d'altérations de la communication interventriculaire, d'un rétrécissement important de l'artère pulmonaire, d'une position anormale de l'aorte et d'un épaississement de la zone ventriculaire droite.
Les personnes atteintes du syndrome de DiGeorge ont généralement une susceptibilité importante à contracter divers types de pathologies, principalement infectieuses (virus, champignons, bactéries, etc.).
Ce fait est dû à la présence d'un dysfonctionnement du système immunitaire, dû à un développement déficient du type et de la production de lymphocytes et de cellules T.
Le système immunitaire est composé d'une grande variété d'organes, de structures, de tissus et de cellules qui, ensemble, nous protègent des agents pathologiques environnementaux et internes..
En ce sens, le syndrome de DiGeorge produit une formation déficiente ou incomplète du thymus, conduisant à des altérations de sa fonctionnalité et de sa localisation finale..
Généralement, l'anomalie la plus importante est l'hypofonctionnalité des lymphocytes T, essentielle dans la production d'immunoglobulines et d'anticorps..
Dans ce cas, les personnes atteintes du syndrome de Digeorge ont généralement des niveaux anormalement bas de concentration de calcium dans le corps et dans la circulation sanguine..
Cette condition médicale dérive fondamentalement de la présence d'anomalies dans les glandes parathyroïdes, en raison d'un sous-développement de ses composants (PrimaryInmune, 2011).
Ces glandes sont situées dans le cou et sont dans une position proche de la thyroïde. Cependant, dans ce cas, ils ont un volume réduit, ce qui aura un impact significatif sur le contrôle du métabolisme et de l'équilibre calcique dans le corps..
Ainsi, dans ce cas, le taux de calcium dans le sang est généralement inférieur à 2,1-8,5 mm / dl, entraînant différentes complications médicales telles que crampes, irritabilité musculaire, engourdissement, sautes d'humeur, déficits cognitifs, etc..
En plus des signes et symptômes décrits ci-dessus, il est possible d'en identifier d'autres liés à la sphère cognitive et intellectuelle des personnes touchées..
Surtout dans les cas diagnostiqués, des difficultés d'apprentissage, un déficit intellectuel modéré, un déficit d'attention, des altérations de l'humeur, des troubles anxieux, entre autres, ont été décrits..
L'origine génétique du syndrome de DiGeorge est associée à la présence d'altérations sur le chromosome 22, en particulier à l'emplacement 22q11.2. Plus précisément, cela est dû à l'absence de séquence d'ADN, composée de 30 à 40 gènes différents..
Si une bonne partie des gènes impliqués n'a pas encore été identifiée en détail, l'absence de ce grand groupe survient dans plus de 90% des cas comme une mutation de novo, tandis qu'environ 7% est due à des facteurs héréditaires..
Pour l'établissement du diagnostic du syndrome de DiGeorge, il est essentiel d'identifier les signes cliniques cardinaux de cette pathologie:
- Défauts faciaux.
- Malformations cardiaques.
- Immunodéficience.
- Hypocalcémie.
En ce sens, parallèlement à l'analyse des antécédents médicaux et à l'examen physique, il est essentiel de réaliser divers tests de laboratoire tels que l'échocardiographie, l'échographie, l'examen immunologique et les études d'analyse sérique..
De plus, un aspect important est l'examen génétique, il est réalisé principalement par hybridation fluorescente in situ (FISH).
Comme nous l'avons souligné dans la description initiale, le traitement est principalement destiné à contrôler et corriger les signes et symptômes provoqués par ce type de maladie..
En cas d'hypocalcémie, elle est généralement traitée par l'administration de suppléments de calcium et / ou de vitamine D..
En revanche, en cas de déficit immunitaire, bien qu'ils tendent à s'améliorer avec l'âge, diverses approches peuvent être utilisées, telles que la transplantation d'une partie du tissu thymique, la thérapie lymphocytaire T ou la greffe de moelle osseuse..
En ce qui concerne les malformations faciales et buccales, des réparations chirurgicales sont généralement utilisées, ce qui améliore l'apparence physique et la fonctionnalité de ces os..
Enfin, dans le cas d'altérations cardiaques, les deux médicaments peuvent être administrés pour leur traitement et leur correction chirurgicale..
Dans la plupart des cas, les personnes touchées atteignent généralement l'âge adulte, cependant, un pourcentage important d'entre elles commencent à développer d'importantes anomalies immunologiques et / ou cardiaques entraînant une mort prématurée, en particulier au cours de la première année de vie..

Personne n'a encore commenté ce post.