le syndrome de Morris, Aussi appelé syndrome d'insensibilité aux androgènes (SIA) ou féminisation testiculaire, c'est une maladie génétique qui affecte le développement sexuel. Les individus qui en souffrent génétiquement sont des hommes, c'est-à-dire qu'ils ont un chromosome X et un chromosome Y dans chaque cellule. Cependant, la forme du corps ne correspond pas à celle dudit sexe.
Pour qu'un phénotype masculin se développe, non seulement certains niveaux d'hormones mâles (testostérone) doivent exister dans le sang; il est également nécessaire que les récepteurs aux androgènes qui les captent fonctionnent correctement.
Ce qui se passe dans ce syndrome, c'est qu'il y a un déficit de ces récepteurs et c'est pourquoi les tissus du corps n'absorbent pas suffisamment de testostérone pour développer une forme masculine..
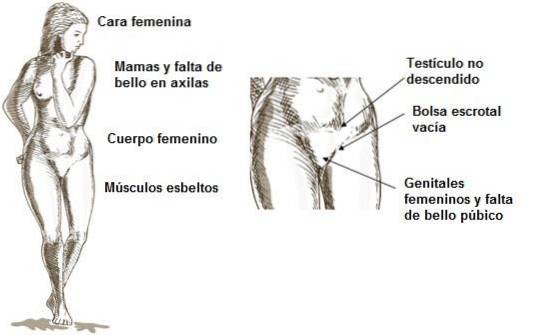
Ainsi, ces personnes naissent avec des organes génitaux féminins apparents et sont généralement élevées comme des filles. Lorsqu'elles atteignent la puberté, les caractéristiques féminines secondaires (élargissement des hanches, voix aiguë, augmentation de la graisse) et les seins se développent. Cependant, ils se rendent compte que la menstruation n'apparaît pas, car ils n'ont pas d'utérus. De plus, ils manquent de poils aux aisselles et au pubis (ou sont absents).
Index des articles
Le syndrome de Morris a été découvert en 1953 par le scientifique et gynécologue John McLean Morris (d'où son nom). Après avoir observé 82 cas (dont deux étaient ses propres patients), il a décrit le "syndrome de féminisation testiculaire".
Morris pensait que cela était dû au fait que les testicules de ces patients produisaient une hormone qui avait un effet féminisant, cependant, on sait maintenant que cela est dû au manque d'action des androgènes dans le corps..
Lorsque la testostérone nécessaire n'est pas absorbée, le corps a tendance à se développer en caractères féminins. Peu importe que les niveaux de testostérone augmentent, le problème réside dans le fait que le corps ne le capte pas. Pour cette raison, le terme «résistance aux androgènes» est plus largement utilisé aujourd'hui..
Nous pouvons également trouver le syndrome de Morris conceptualisé comme pseudohermaphrodisme masculin.
Selon Borrego López, Varona Sánchez, Areces Delgado et Formoso Martín (2012); On estime que le syndrome de Morris survient chez un nouveau-né de sexe masculin sur 20 000 à 64 000. Le chiffre pourrait même être plus élevé si les cas non encore diagnostiqués ou qui ne nécessitent pas d'assistance médicale sont comptés.
Le syndrome de Morris est considéré comme la troisième cause d'aménorrhée après la dysgénésie gonadique et l'absence du vagin à la naissance.
Il n'y a pas de degré unique d'insensibilité aux androgènes, mais les caractéristiques du syndrome dépendent du niveau de déficit des récepteurs aux androgènes.
Ainsi, il peut y avoir moins de récepteurs de la dihydrotestostérone que d'habitude et recevoir moins de testostérone que nécessaire, ou il peut y avoir des cas où le déficit en récepteurs est total..
Les trois types classiques d'insensibilité aux androgènes (AIS) sont:
- Syndrome d'insensibilité légère aux androgènes: organes génitaux externes masculins.
- Syndrome d'insensibilité partielle aux androgènes: organes génitaux partiellement masculinisés.
- Syndrome d'insensibilité complète aux androgènes: organes génitaux féminins.
Le syndrome de Morris fait partie de ce dernier, car il existe une résistance totale aux androgènes chez les patients nés avec des organes génitaux externes féminins..
Dans les formes incomplètes, différents niveaux de traits masculins et féminins peuvent apparaître tels que la clitoromégalie (clitoris plus grand que la normale) ou la fermeture partielle du vagin externe..
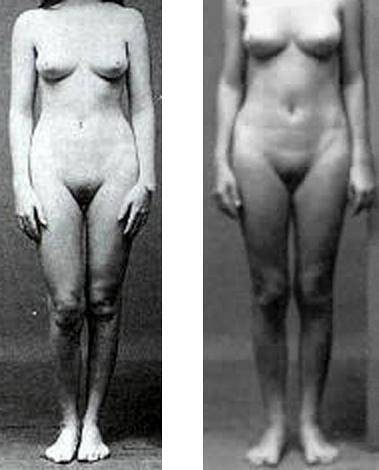
Les personnes atteintes du syndrome de Morris ne manifesteront pas de symptômes dans l'enfance. En fait, la plupart reçoivent le diagnostic lorsqu'ils vont chez le spécialiste avec la raison pour laquelle la menstruation n'apparaît pas.

Les caractéristiques généralement présentes sont les suivantes:
- 46 caryotype XY, qui est associé au sexe masculin.
- Les organes génitaux externes ont une apparence féminine, bien qu'avec une hypoplasie des grandes lèvres et des mineures. Cela signifie que les lèvres ne sont pas complètement développées, étant plus petites..
- Malgré des organes génitaux externes normaux, le vagin est peu profond et se termine par un cul-de-sac aveugle. Autrement dit, il n'est pas connecté à l'utérus car le plus souvent il n'a pas été formé.
- Les ovaires sont parfois absents ou atrophiés.
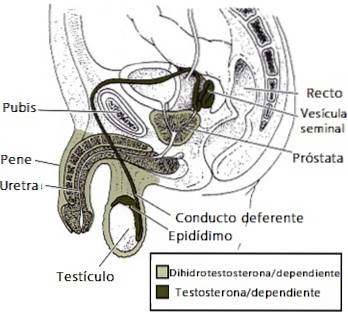
- Ils ont généralement des testicules non descendus qui se trouvent dans l'aine, l'abdomen ou les grandes lèvres. Parfois, les testicules sont à l'intérieur d'une hernie inguinale qui peut être ressentie à l'examen physique.
Ces testicules sont normaux avant la puberté, mais après la puberté, les tubules séminifères sont plus petits et la spermatogenèse ne se produit pas..
- À la puberté, les caractéristiques sexuelles secondaires féminines normales se développent pour atteindre une apparence féminine complète. Cela est dû à l'action de l'estradiol, une hormone sexuelle féminine produite dans diverses parties du corps..
Une caractéristique distinctive du syndrome est qu'ils n'ont pas de poils aux aisselles ou au pubis, ou c'est très peu.
- Absence de ménarche (la première menstruation).
- Les taux de testostérone dans le sang sont typiques chez les hommes, mais comme les récepteurs androgènes ne fonctionnent pas correctement, les hormones mâles ne peuvent pas faire leur travail..
- Bien sûr, cette maladie provoque l'infertilité.
- S'il n'est pas intervenu, les difficultés dans les relations sexuelles sont fréquentes, telles que des problèmes de pénétration et de dyspareunie (douleur).
- Une diminution de la densité osseuse a été observée chez ces patients, ce qui peut être dû à l'influence des androgènes..
- Si les testicules ne sont pas retirés, le risque de tumeurs germinales malignes augmente avec l'âge. Dans une étude, le risque a été estimé à 3,6% à 25 ans et à 33% à 50 ans (Manuel, Katayama & Jones, 1976).
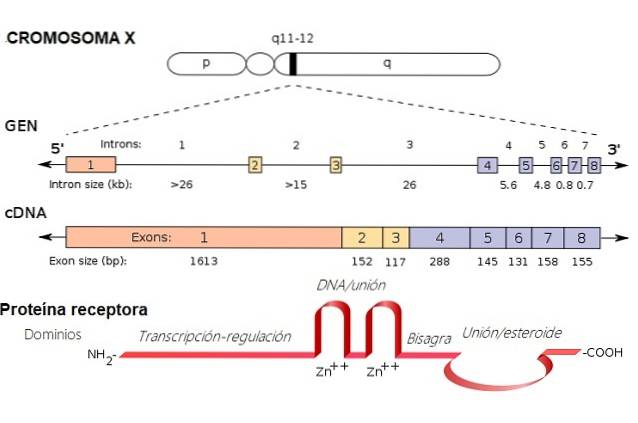
Le syndrome de Morris est une maladie héréditaire, avec un modèle récessif lié à X. Cela signifie que le gène muté qui cause le syndrome est situé sur le chromosome X..
Il survient plus fréquemment chez les hommes que chez les femmes, car les femmes ont besoin de mutations sur les deux chromosomes (XX) pour présenter le trouble. Au lieu de cela, les hommes peuvent le développer avec une mutation sur leur chromosome X (ils n'en ont qu'une).
Ainsi, les femmes peuvent être porteuses du gène muté, mais pas avoir le syndrome. En fait, il semble qu'environ les deux tiers de tous les cas de résistance aux androgènes sont hérités de mères qui ont une copie modifiée du gène sur l'un de leurs deux chromosomes X..
Les autres cas sont dus à une nouvelle mutation qui semble survenir dans l'ovule maternel au moment de la conception ou au cours du développement du fœtus (Genetics Home Reference, 2016).
Les mutations de ce syndrome sont localisées dans le gène AR, qui est responsable de l'envoi des instructions pour le développement des protéines AR (Androgen Receptor). Ce sont ceux qui médiatisent les effets des androgènes dans le corps.
Les récepteurs capturent les hormones sexuelles mâles telles que la testostérone, les envoyant aux différentes cellules pour un développement masculin normal..
Lorsque ce gène est modifié, comme cela se produit dans le syndrome de Morris, des déficits à la fois quantitatifs (nombre de récepteurs) et qualitatifs (récepteurs anormaux ou défectueux) des récepteurs androgènes peuvent survenir..
De cette manière, les cellules ne répondent pas aux androgènes, c'est-à-dire que les hormones mâles n'agissent pas. Par conséquent, le développement du pénis et d'autres caractéristiques typiques de l'homme est entravé, et un développement féminin est cédé..
Plus précisément, la testostérone qui existe chez ces individus est aromatisée (transformée par l'enzyme aromatase) en œstrogène, une hormone sexuelle qui est à l'origine de l'apparition féminine dans le syndrome de Morris..
Certains traits masculins se développent parce qu'ils ne dépendent pas des androgènes. Par exemple, les testicules sont formés en raison du gène SRY présent sur le chromosome Y..
Le diagnostic du syndrome de Morris est généralement posé après la puberté, car ces patients ne remarquent généralement aucun symptôme avant celle-ci. Cependant, c'est un syndrome difficile à diagnostiquer, car l'apparence est totalement féminine et jusqu'à ce qu'une scintigraphie de la région pelvienne ou une étude chromosomique soit effectuée, le problème n'est pas détecté..
Si le syndrome de Morris est suspecté, le spécialiste établira un diagnostic basé sur:
- Antécédents cliniques complets du patient, étant important qu'il n'ait pas présenté de menstruation.
- Examen physique qui peut être basé sur l'échelle de Tanner, qui reflète le niveau de maturation sexuelle. Dans ce syndrome, il devrait être normal dans les seins, mais moins dans les organes génitaux et les cheveux dans les aisselles et le pubis.
L'échelle de Quigley, qui mesure le degré de masculinité ou de féminité des organes génitaux, peut également être utilisée. Grâce à cet indice, il est également possible de distinguer les différents types d'insensibilité aux androgènes.
- Échographie gynécologique: les images des organes génitaux internes sont obtenues par ondes sonores. L'utérus ou les ovaires ne sont souvent pas vus, mais des testicules peuvent être présents dans une zone voisine. Le vagin est généralement plus court que la longueur normale.
- Études hormonales: grâce à un test sanguin, il est pratique d'explorer les niveaux de testostérone (dans le syndrome de Morris, ils sont élevés et similaires aux niveaux masculins), les hormones folliculo-stimulantes (FSH), les hormones lutéinisantes (LH) ou l'estradiol (E2).
- Etude chromosomique: elles peuvent être effectuées à travers un échantillon de sang, une biopsie cutanée ou tout autre échantillon de tissu. Dans ce syndrome, le résultat devrait être un caryotype 46 XY.
Dans l'histoire, il y a eu des conflits pour décider quand et comment révéler un diagnostic de syndrome de Morris à la personne touchée. Dans les temps anciens, il était souvent caché par les médecins et les proches, mais cela a évidemment un impact encore plus négatif sur la personne..
Malgré le dilemme qu'il génère, nous devons essayer de faire en sorte que le patient reçoive l'information dans un environnement empathique et détendu, répondant à toutes ses préoccupations.
Il n'existe actuellement aucune méthode pour corriger la déficience des récepteurs aux androgènes présente dans le syndrome de Morris. Mais il existe d'autres interventions qui peuvent être effectuées:
Avant d'envisager une intervention chirurgicale, une tentative est faite pour augmenter la taille du vagin en utilisant des méthodes de dilatation. Ceci est recommandé après la puberté.
Le vagin étant élastique, cette thérapie consiste en l'introduction et la rotation d'un objet de forme phallique plusieurs fois par semaine pendant quelques minutes, ceci étant progressif.
Les testicules doivent être retirés chez les patients atteints du syndrome de Morris, car ils ont tendance à développer des tumeurs malignes (carcinomes) s'ils ne sont pas retirés. Il est essentiel pour un bon pronostic qu'ils soient extraits le plus tôt possible.
Il est essentiel chez ces patients qu'ils reçoivent un traitement psychologique, car ce syndrome peut provoquer une insatisfaction importante vis-à-vis du corps lui-même. Grâce à ce type d'intervention, la personne pourra accepter sa situation et mener une vie la plus satisfaisante possible, en évitant l'isolement social.
Vous pouvez même travailler sur les liens familiaux, afin que la famille soutienne et contribue au bien-être du patient..
Pour la diminution de la densité osseuse typique de ces patients, des suppléments de calcium et de vitamine D. L'exercice peut également être très bénéfique..
Dans les cas plus graves, l'utilisation de bisphosphonates, médicaments qui inhibent la résorption osseuse, peut être recommandée..
Si les méthodes de dilatation n'ont pas été efficaces, la reconstruction d'un vagin fonctionnel peut être une alternative. La procédure est appelée néovaginoplastie, et pour la reconstruction, des greffes cutanées du patient de l'intestin ou de la muqueuse buccale sont utilisées.
Après la chirurgie, des méthodes de dilatation seront également nécessaires..
Des tentatives ont été faites pour administrer des œstrogènes à ces patients pour atténuer le manque de densité osseuse, mais cela ne semble pas avoir l'effet souhaité sur tout le monde..
En revanche, des androgènes ont été administrés après l'ablation des testicules (car il y a une baisse significative de leur taux). Les androgènes semblent maintenir un sentiment de bien-être chez les patients.


Personne n'a encore commenté ce post.