
le Syndrome de Pallister-Killian, également connue sous le nom de tétrasomie 12, c'est une maladie rare d'origine génétique qui se caractérise par un large spectre d'implication multi-organes.
Cliniquement, cette pathologie est définie par une déficience intellectuelle, un retard psychomoteur, une hypotonie musculaire, un phénotype facial atypique, des anomalies pigmentaires de la peau et une alopécie. En outre, d'autres types de complications médicales liées à des malformations dans différents systèmes corporels ou à des convulsions peuvent également apparaître..
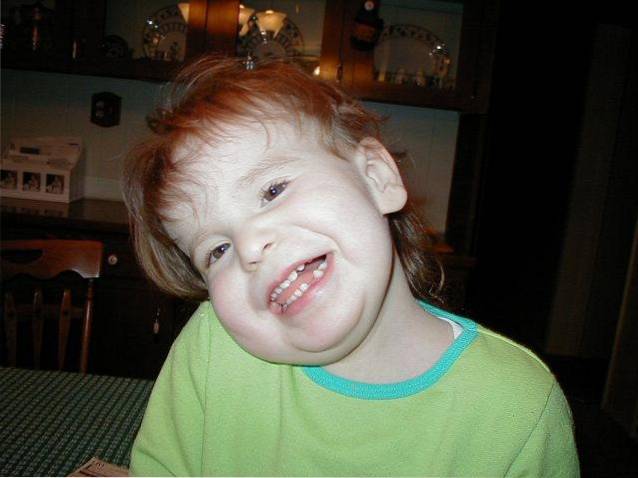
L'origine étiologique de cette maladie est associée à une maladie génétique répartie en mosaïque. Plus précisément, cela est dû à la présence d'un chromosome 12 supplémentaire dans certaines cellules du corps.
Le diagnostic du syndrome de Pallister-Killiam peut être posé à la fois avant et après la naissance. L'objectif principal est l'identification des caractéristiques cliniques et l'utilisation d'une étude génétique de confirmation..
Ce syndrome a un taux de mortalité élevé. Cependant, l'approche médicale pharmacologique et le traitement de rééducation peuvent apporter des bénéfices importants sur la qualité de vie et l'état clinique des personnes touchées..
Index des articles
Cette maladie a été initialement décrite par Pallister en 1977. Dans les premières publications, ce chercheur rapportait deux cas de patients adultes dont l'évolution était caractérisée par divers constats: convulsions, hypotonie musculaire, déficit intellectuel, malformations musculo-squelettiques et organiques, configuration du visage rugueuse et couleur de la peau changements.
En parallèle, Teschler-Nicola et Killiam en 1981 ont décrit ce même tableau clinique chez une fillette de trois ans.
Par conséquent, dans les premiers rapports cliniques, une référence générale a été faite à une condition médicale caractérisée par la combinaison de crises d'épilepsie, de déficience intellectuelle et d'un phénotype physique caractéristique..
De plus, déjà en 1985, Gilgenkratz a pu identifier dans le premier cas pendant la phase de gestation, chose courante aujourd'hui grâce aux techniques de diagnostic modernes.
Le syndrome de Pallister-Killiam est un type de maladie de la mosaïque génétique. Dans ce cas, l'altération chromosomique n'affecte que certaines cellules du corps. Une large implication de différents systèmes corporels et organismes est identifiée.
Elle se caractérise principalement par une déficience intellectuelle, une hypotonie musculaire, le développement de traits distinctifs du visage, une altération de la pigmentation de la peau ou de la croissance des cheveux, entre autres altérations congénitales..
De plus, le syndrome de Pallister-Kiliam est une maladie rare d'origine congénitale qui peut recevoir un grand nombre de noms dans la littérature médicale:
Les chiffres de prévalence du syndrome de Pallister-Killiam ne sont pas connus avec précision. Peu de diagnostics définitifs ont été posés et la plupart n'ont pas été publiés dans la littérature médicale.
Ainsi, tous les auteurs et institutions définissent ce syndrome comme une pathologie génétique rare ou peu fréquente dans la population générale..
Il y a environ 15 ans, le syndrome de Pallister-Killiam avait été identifié dans une centaine de cas dans le monde. Actuellement, ce chiffre a dépassé les 200 touchés.
Les enquêtes épidémiologiques ont estimé l'incidence de cette maladie à environ 5,1 cas par million de nouveau-nés, bien que des auteurs tels que Toledo-Bravo de la Laguna et des collaborateurs la situent à 1/25 000.
Une prévalence plus élevée associée aux caractéristiques sociodémographiques des personnes touchées n'a pas été identifiée. Le syndrome de Pallister-Killian peut apparaître dans n'importe quel sexe ou groupe technique et / ou racial.
Une grande variété de signes et de symptômes peuvent être identifiés au cours de l'évolution clinique du syndrome de Pallister-Killian. Tous associés à des anomalies cranio-faciales et / ou musculo-squelettiques et à des altérations cognitives.
Le développement de malformations cranio-faciales de la phase de gestation à la croissance postnatale et infantile constitue l'un des signes médicaux les plus caractéristiques du syndrome de Pallister-Killiam..
Les signes et symptômes les plus courants incluent des anomalies dans les différentes structures crâniennes et faciales qui conduiront à une apparence rugueuse et atypique:
Bien que moins significatives que les altérations faciales, il est très fréquent d'observer plusieurs anomalies musculo-squelettiques chez les patients atteints du syndrome de Pallister:
Les anomalies liées à la structure musculaire et à la mobilité sont une autre des caractéristiques cliniques cardinales du syndrome de Pallister-Killian:
L'hypotonie musculaire fait référence à l'identification d'un tonus ou d'une tension musculaire anormalement réduite. Au niveau visuel, la flaccidité et la labilité peuvent être observées dans différents groupes musculaires, particulièrement accentuées dans les extrémités..
Ainsi, la pathologie musculaire et squelettique entraînera un retard important dans l'acquisition de différentes habiletés motrices, aussi bien en période néonatale que infantile..
Bien que les périodes de développement varient selon les personnes concernées, le calendrier le plus courant comprend les jalons suivants:
Une autre des zones fortement touchées est le système nerveux. Dans la plupart des cas, les signes et symptômes sont principalement liés à des convulsions et à une déficience intellectuelle:
Bien qu'elles soient moins fréquentes, d'autres types de complications médicales peuvent également apparaître:
L'origine du syndrome de Pallister-Killian est associée à une anomalie génétique de la mosaïque sur le chromosome 12. Elle n'affecte que le matériel génétique de certaines cellules du corps..
Les chromosomes font partie du noyau de toutes les cellules présentes dans le corps humain. Ils sont constitués d'une grande variété de composants biochimiques et contiennent les informations génétiques de chaque individu..
Les êtres humains ont 46 chromosomes différents, organisés par paires et numérotés de 1 à 23. De plus, au niveau individuel, chaque chromosome a une courte zone ou un bras appelé "p" et un long appelé "q".
L'anomalie affecte le chromosome 12 et se traduit par la présence d'un chromosome avec une structure anormale, appelée isochromosome.
Ainsi, ce chromosome a tendance à avoir deux bras courts au lieu d'un de chaque configuration p (court) et long (q)..
En conséquence, la présence de matériel génétique supplémentaire et / ou anormal modifiera le déroulement normal et efficace du développement physique et cognitif de la personne affectée, donnant lieu aux caractéristiques cliniques du syndrome de Pallister-Killian..
Le syndrome de Pallister-Killian peut être identifié pendant la grossesse ou au stade postnatal, sur la base des caractéristiques cliniques et des résultats de différents tests de laboratoire..
Pendant la grossesse, les tests les plus couramment utilisés sont les échographies, l'amniocentèse ou le prélèvement des villosités choriales. En ce sens, l'analyse du matériel génétique de l'embryon peut nous offrir une confirmation de cette pathologie, à travers l'identification d'anomalies compatibles..
En revanche, si le diagnostic est posé après la naissance, il est indispensable:
Aucun traitement spécifique n'a été conçu pour traiter les personnes atteintes du syndrome de Pallister-Killian..
Ce syndrome est généralement associé à un mauvais pronostic neurologique et à des taux de mortalité élevés. Cependant, un traitement de réadaptation, une éducation spécialisée et une ergothérapie peuvent offrir un bon pronostic fonctionnel et une amélioration de la qualité de vie des personnes touchées..
Par exemple, Méndez et son équipe de travail (2013) décrivent un cas de traitement de réadaptation caractérisé par:


Personne n'a encore commenté ce post.