
le Syndrome de Williams il s'agit d'un trouble du développement d'origine génétique associé à un profil caractéristique de déficiences physiques et cognitives. Plus précisément au niveau clinique, elle est caractérisée par 4 points cardinaux: 1) traits et caractéristiques faciales atypiques, 2) retard généralisé du développement psychomoteur et profil neurocognitif spécifique, 3) altérations cardiovasculaires et t) possibilité de développer une hypercalcémie infantile.
Bien que le syndrome de Williams soit considéré comme une maladie rare, il y a des milliers de personnes touchées dans le monde. En ce qui concerne le diagnostic, l'examen clinique fournit généralement les résultats nécessaires à son établissement, cependant, pour écarter d'autres pathologies et faux positifs, une étude génétique est généralement initiée par diverses techniques..
D'un autre côté, il n'y a ni remède pour le syndrome de Williams ni protocole de traitement standard, de sorte que la plupart des interventions thérapeutiques tenteront de réguler les complications médicales. De plus, il sera essentiel d'inclure dans les interventions des programmes de soins précoces, une éducation spécialisée individualisée et une stimulation neuropsychologique..
Index des articles
Le syndrome de Williams est un trouble du développement qui peut affecter de manière significative différentes zones.
Généralement, cette pathologie se caractérise par la présence de traits du visage atypiques ou d'altérations cardiovasculaires, une déficience intellectuelle modérée, des problèmes d'apprentissage et des traits de personnalité distinctifs..
Ainsi, le premier patient atteint du syndrome de Williams a été décrit par le Dr Guido Fanconi, dans un rapport clinique de 1952. Cependant, c'est le cardiologue Joseph Williams qui en 1961 a précisément identifié cette pathologie, tout en étant décrite par l'Allemand Beuren.
Pour cette raison, le syndrome de Williams tire son nom des deux auteurs (syndrome de Williams-Beuren), ou simplement du premier.
Malgré le fait que, jusqu'à il y a quelques années, l'identification de la pathologie était effectuée sur la base des caractéristiques phénotypiques, en 1993 Edward et al.ont trouvé une anomalie génétique dans le chromosome 7q 11.23 comme cause étiologique..
Bien que le syndrome de Williams soit associé à une grande variété de complications médicales secondaires, il n'a pas un taux de mortalité élevé. Dans de nombreux cas, les personnes touchées sont capables d'atteindre un niveau fonctionnel indépendant.
Le syndrome de Williams est considéré comme une maladie génétique rare ou rare.
La Williams Syndrome Association, entre autres institutions, a estimé que le syndrome de Williams a une prévalence d'environ 1 cas pour 10 000 personnes dans le monde. Plus précisément, il a été identifié qu'aux États-Unis, il peut y avoir environ 20000 ou 30000 touchés.
En ce qui concerne la répartition de la pathologie par sexe, il n'y a pas de données récentes indiquant une prévalence plus élevée dans aucune d'entre elles, de plus, aucune différence n'a été identifiée entre les régions géographiques ou les groupes ethniques..
D'autre part, nous savons également que le syndrome de Williams est une condition médicale sporadique, bien que certains cas de transmission familiale aient été décrits..
Le syndrome de Williams, comme d'autres pathologies d'origine génétique, a une évolution clinique caractérisée par une implication multisystémique.
De nombreux auteurs, tels que González Fernández et Uyaguari Quezada, décrivent le spectre clinique du syndrome de Williams catégorisé en plusieurs domaines: caractéristiques biomédicales, caractéristiques psychomotrices et cognitives, caractéristiques psychologiques et comportementales, entre autres..
L'affectation physique présente dans le syndrome de Wiliams est diverse, parmi les résultats cliniques les plus fréquents que nous pouvons observer:
Un développement retardé ou ralenti peut déjà être détecté pendant la grossesse. Les enfants atteints du syndrome de Williams naissent souvent avec un poids et une taille faibles. De plus, une fois le stade adulte atteint, la hauteur totale est généralement inférieure à celle de la population générale, environ 10-15 cm.
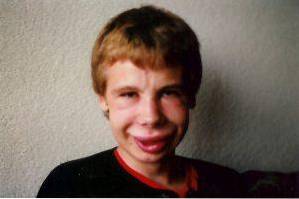
Les altérations du visage sont l'un des signes cliniques les plus caractéristiques de ce syndrome. Chez les personnes atteintes, nous pouvons observer un front significativement étroit, des plis cutanés marqués dans la fissure palpébrale, un strabisme, un iris étoilé, un nez court et aplati, des pommettes proéminentes et un menton plus petit que d'habitude..
Dans le cas d'altérations liées au développement des muscles et des os, il est possible d'observer la présence d'une diminution du tonus musculaire et de la force, la laxité articulaire, la scoliose, les contractures, entre autres. Visuellement, une posture caractérisée par des épaules tombantes et des membres inférieurs semi-fléchis peut être observée..
Bien qu'il n'y ait généralement pas d'anomalies ou de malformations significatives dans le pavillon auditif, dans tous les cas, une augmentation de la sensibilité auditive se développe. Les personnes touchées ont tendance à percevoir ou à ressentir certains sons comme étant ennuyeux ou douloureux.
La peau a généralement peu d'élasticité, il est donc possible d'observer les premiers signes du vieillissement. De plus, des hernies peuvent se développer, en particulier dans la région inguinale et ombilicale..
Les différentes anomalies du cœur et des vaisseaux sanguins constituent la complication médicale la plus importante, car elles peuvent mettre en danger la survie de la personne touchée.
Parmi les anomalies cardiovasculaires, certaines des plus courantes sont la sténose aortique supravalvulaire, la sténose de la branche pulmonaire, la sténose de la valve aortique. Toutes ces altérations, au niveau clinique, peuvent toucher d'autres territoires vasculaires et même le cerveau, du fait du développement de l'hypertension artérielle.
Les anomalies liées à la fonction rénale et à la vessie sont très fréquentes. De plus, une accumulation de calcium (néphrocalcinose), une urgence urinaire ou une énurésie nocturne peuvent également être détectées.
Au niveau cognitif, les caractéristiques les plus significatives sont constituées par un retard généralisé dans l'acquisition de la motricité, un retard intellectuel modéré et diverses altérations liées à la perception visuelle.
Différentes altérations liées aux problèmes d'équilibre et de coordination sont décrites, qui sont principalement dues à la présence d'anomalies musculo-squelettiques et qui entraîneront, entre autres, un retard dans l'acquisition de la démarche, la motricité finale, etc..
Il est possible de trouver un retard mental modéré, le QI typique des personnes touchées oscille généralement entre 60 et 70. En ce qui concerne les zones spécifiques touchées, il existe une asymétrie claire: en plus de la coordination psychomotrice, de la perception et de l'intégration visuelle, il être clairement touchés, alors que des domaines tels que la langue sont généralement plus développés.
Dans les stades les plus initiaux, il y a généralement un retard dans l'acquisition des compétences linguistiques, cependant, il récupère généralement environ 3-4 ans. Les enfants atteints du syndrome de Williams ont tendance à avoir une bonne communication expressive, sont capables d'utiliser un vocabulaire contextualisé, une grammaire correcte, un contact visuel, des expressions faciales, etc..
L'une des découvertes les plus significatives dans le syndrome de Williams est le comportement social exceptionnel des personnes touchées. Bien que dans certains cas des crises d'anxiété ou des inquiétudes excessives puissent survenir, elles sont très empathiques et sensibles.
Les recherches les plus récentes ont indiqué que la cause du syndrome de Williams se trouve dans diverses altérations génétiques sur le chromosome 7. Les chromosomes portent l'information génétique de chaque personne et sont situés dans le noyau des cellules du corps..
Chez l'homme, nous pouvons trouver 46 chromosomes répartis par paires. Celles-ci sont numérotées de 1 à 23, à l'exception de la dernière paire constituée des chromosomes sexuels, appelée XX pour les femmes, XY pour les hommes. Ainsi, dans chaque chromosome, il peut y avoir une infinité de gènes.
Plus précisément, le processus anormal identifié dans le syndrome de Williams est une microcélection ou une dégradation d'une molécule d'ADN qui confirme ce chromosome. Normalement, ce type d'erreur a lieu dans la phase de développement des gamètes mâles ou femelles..
Des anomalies génétiques se trouvent dans la zone 7q11.23, dans laquelle plus de 25 gènes différents liés au schéma clinique caractéristique de cette pathologie ont été identifiés..
Certains des gènes, tels que Clip2, ELN, GTF21, GTF2IRD1 ou LIMK1, sont absents chez les personnes touchées. La perte d'ELN est liée à des anomalies du tissu conjonctif, de la peau et du système cardiovasculaire.
D'autre part, certaines recherches indiquent que la perte des gènes Clip2, GTF2I, GTF2IRD1 et LIMK1 peut expliquer des altérations des processus visuopérceptifs, du phénotype comportemental ou des déficits cognitifs..
De plus, spécifiquement, le gène GTF2IRD1 semble jouer un rôle de premier plan dans le développement de traits atypiques du visage. De son côté, le gène NCF1 semble être lié à un risque élevé de développer une hypertension.
Jusqu'à ces dernières années, le diagnostic du syndrome de Williams était exclusivement basé sur l'observation des caractéristiques phénotypiques (altérations du visage, déficience intellectuelle, déficits cognitifs spécifiques, entre autres).
Cependant, à l'heure actuelle, le diagnostic du syndrome de Williams se fait généralement en deux étapes: l'analyse des résultats cliniques et les études génétiques de confirmation. Ainsi, le diagnostic clinique comprend généralement:
- Examen et évaluation physique et neurologique.
- Analyse des paramètres de croissance.
- Examen du système cardiorespiratoire.
- Examen néphrourologique.
- Analyse des taux de calcium dans l'urine et le sang.
- Analyse ophtalmologique.
D'autre part, l'analyse génétique est utilisée pour confirmer la présence d'altérations génétiques compatibles avec le syndrome de Williams, parmi les tests les plus courants est la technique d'hybridation fluorescente in situ (FIHS)..
Après l'extraction d'un échantillon sanguin, la technique d'hybridation in situ est réalisée en marquant des sondes d'ADN qui sont détectées sous une lumière fluorescente..
Il n'y a pas de traitement spécifique pour le syndrome de Williams, cependant, cette pathologie est associée à de multiples complications dans différents organes, les interventions médicales seront donc orientées vers le traitement de ces derniers..
Les auteurs González Fernández et Uyaguari Quezada soulignent que toutes les interventions doivent avoir un caractère multidisciplinaire marqué, permettant le traitement de la variété symptomatique caractéristique de ce syndrome. En outre, ils soulignent également diverses mesures thérapeutiques en fonction de la zone touchée:
Dans ce cas, les complications médicales telles que les altérations cardiaques ou les malformations musculo-squelettiques nécessitent généralement un traitement basé principalement sur l'administration de médicaments et d'interventions chirurgicales. Des professionnels de la santé de différents domaines (pédiatres, cardiologues, ophtalmologistes, etc.) participent généralement au traitement des symptômes physiques..
Les déficits cognitifs tels que l'altération de la perception visuelle ou le retard linguistique doivent être traités dès les premiers stades. La stimulation cognitive et la rééducation seront un facteur déterminant pour parvenir à une vie autonome à l'âge adulte.
Bien que les personnes atteintes du syndrome de Williams présentent généralement un bon fonctionnement social, elles ont parfois tendance à montrer des comportements excessivement anxieux et à développer des comportements ou des phobies persévérants..
Par conséquent, dans ces cas, il sera essentiel de mettre en œuvre une approche psychologique, à travers diverses stratégies efficaces pour minimiser ces problèmes ou difficultés..



Personne n'a encore commenté ce post.