
le Syndrome d'Apert ou l'acrocephalosyndactylie de type I (ACS1) est une pathologie d'origine génétique qui se caractérise par la présence de différentes altérations et malformations au niveau du crâne, du visage et des extrémités.
Au niveau clinique, le syndrome d'Apert se caractérise par la présence ou le développement d'un crâne pointu ou allongé, une zone faciale enfoncée avec une altération de la projection des dents, la fusion et la fermeture des os et des articulations des doigts, un retard mental variable, des troubles du langage , etc..
Bien que cette pathologie puisse être héréditaire, le syndrome d'Apert survient dans la plupart des cas sans la présence d'antécédents familiaux, essentiellement dus à une mutation de novo pendant la phase de gestation..
Les mécanismes génétiques à l'origine du syndrome d'Apert ne sont pas exactement connus. Actuellement, diverses altérations génétiques susceptibles de produire cette pathologie ont été identifiées, essentiellement liées à des mutations du gène FGFR2.
D'autre part, le diagnostic du syndrome d'Apert commence généralement par une suspicion clinique dans la période prénatale après l'identification d'anomalies dans les échographies de routine et est confirmé par la conduite d'une étude génétique..
Concernant le traitement, il n'y a pas de type d'intervention curative pour le syndrome d'Apert. Cependant, tout au long de l'histoire de cette pathologie, diverses interventions spécifiques ont été conçues qui incluent généralement la neurochirurgie, la chirurgie craniofaciale, la chirurgie maxillo-faciale, le traitement médicamenteux, la physiothérapie, l'intervention psychologique et neuropsychologique, entre autres..
Index des articles
Le syndrome d'Apert est une pathologie génétique caractérisée par la présence de différentes malformations squelettiques au niveau du crâne, du visage et / ou des membres.
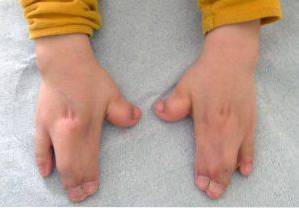
L'altération essentielle du syndrome d'Apert est constituée par une fermeture prématurée ou précoce des fissures crâniennes, qui provoque une croissance anormale du reste des structures du visage et du crâne. En plus de ceux-ci, des malformations peuvent également apparaître dans les membres supérieurs et inférieurs, comme la fusion des doigts et des orteils..
D'autre part, les capacités cognitives des personnes atteintes du syndrome d'Apert peuvent également être affectées, avec une gravité variable allant de légère à modérée..
Bien que Baumgartner (1842) et Wheaton (1894) aient fait les premières mentions de cette condition médicale, ce n'est qu'en 1906, lorsque le médecin spécialiste français Eugène Apert, décrit avec précision ce syndrome et publie le premier rapport clinique.
Dans sa publication, Eugene Apert, décrit un ensemble de nouveaux cas de patients atteints d'un schéma malformatif bien défini et caractérisés par les signes et symptômes caractéristiques de cette pathologie..
Ainsi, ce n'est qu'en 1995 que les facteurs génétiques étiologiques du syndrome d'Apert ont été identifiés. Plus précisément, Wilkie et al.ont décrit la présence de deux mutations dans le gène FGFR2 chez environ 40 patients atteints..
De plus, le syndrome d'Apert est une affection médicale classée dans les maladies ou pathologies caractérisées par la présentation d'une craniosynostose (fermeture prématurée des sutures crâniennes).
Les autres pathologies appartenant à ce groupe sont le syndrome de Pfeiffer, le syndrome de Crouzon, le syndrome de Saethre-Chotzcen et le syndrome de Carpenter..
Le syndrome d'Apert est considéré comme une pathologie rare ou peu fréquente, c'est-à-dire qu'il a une prévalence de moins d'un cas pour 15 000 habitants de la population générale..
Plus précisément, le syndrome d'Apert survient autour d'une personne pour 160000 à 200000 naissances et, en outre, il existe une probabilité de 50% de transmettre cette pathologie au niveau héréditaire..
En outre, en ce qui concerne la répartition par sexe, une prévalence plus élevée chez les hommes ou les femmes n'a pas été identifiée, ni associée à des groupes ethniques ou à des lieux géographiques particuliers..
Actuellement, et étant donné que le syndrome d'Apert a été identifié vers 1984, dans des rapports cliniques et dans la littérature médicale qui ont publié plus de 300 cas de cette pathologie.
Les manifestations cliniques du syndrome d'Apert comprennent généralement une malformation ou un développement incomplet de la structure crânienne, un phénotype ou un motif facial atypique et des modifications squelettiques des extrémités..
Dans le cas du syndrome d'Apert, l'atteinte centrale est liée à la formation et à la fermeture de la structure osseuse du crâne. Au cours du développement embryonnaire, un processus appelé crénéosynostose se produit, caractérisé par une fermeture prématurée des sutures crâniennes.
Les fissures ou sutures crâniennes sont un type de bandes de tissu fibreux qui ont pour objectif fondamental de relier les os qui composent le crâne (frontal, occipital, pariétal et temporal).
Pendant la phase de gestation et la période postnatale précoce, la structure osseuse qui compose le crâne est maintenue ensemble grâce à ces tissus fibreux et élastiques.
Normalement, les os crâniens ne fusionnent que vers 12 à 18 mois. La présence de points faibles ou d'espaces entre les os crâniens fait partie du développement normal de l'enfant.
Ainsi, pendant toute la durée de l'enfance, ces sutures ou régions flexibles permettent au cerveau de se développer de manière accélérée et, en plus, le protègent des chocs..
Ainsi, dans le syndrome d'Apert, la fermeture prématurée de ces sutures crâniennes et des os crâniens rend impossible le développement normal de la croissance crânienne et cérébrale..
Par conséquent, les signes et symptômes les plus courants du syndrome d'Apert peuvent inclure:
Ces types d'altérations affectent principalement les membres supérieurs et inférieurs, normalement la fusion et le développement des doigts..
En plus de ceux-ci, il est également possible d'observer d'autres signes cliniques au niveau musculo-squelettique, raccourcissement de divers os (radius, humérus, fémur), hypoplasie de l'omoplate ou du bassin, fusion des vertèbres cervicales.
En conséquence, de nombreux malades présenteront une mobilité articulaire réduite et, par conséquent, peuvent développer diverses difficultés pour l'acquisition de la motricité globale et fine..
Ces types d'anomalies sont très hétérogènes et variables selon les personnes touchées, cependant, certaines des plus courantes ont été identifiées:
L'altération étiologique de cette pathologie peut conduire au développement de lésions ou de pathologies secondaires au niveau morphologique et structurel dans diverses zones du corps, dont certaines comprennent:
Malgré le fait que dans de nombreux cas, il est possible d'observer la présence d'une altération générale des fonctions cognitives et du niveau intellectuel, le retard mental n'est pas clairement présent dans tous les cas de syndrome d'Apert..
De plus, dans les cas où il y a une altération du niveau intellectuel, celle-ci peut être variable, sur une échelle de légère à modérée.
En revanche, dans le domaine linguistique, le développement de divers déficits est fréquent, principalement lié à l'articulation des sons à la suite de malformations mandibulaires et orales..
Le syndrome d'Apert est dû à la présence d'une mutation spécifique dans le gène FGFR2. Des études expérimentales ont indiqué que ce gène est responsable de la production d'une protéine, appelée récepteur 2 du facteur de croissance des fibroblastes.
Parmi les fonctions de ce facteur, il décrit l'envoi de différents signaux chimiques à des cellules immatures pour provoquer leur transformation et leur différenciation en cellules osseuses au cours de la phase de développement fœtal ou prénatal..
Par conséquent, la présence de mutations dans le gène FGFR2 altère le fonctionnement de cette protéine et, par conséquent, peut provoquer la fusion précoce des os du crâne, de la main et des pieds..
Une bonne partie des caractéristiques cliniques du syndrome d'Apert peut être identifiée pendant la grossesse, en particulier lors des examens échographiques de la grossesse et du développement fœtal..
Ainsi, en cas de suspicion clinique, une étude génétique est relancée pour identifier la présence d'une mutation génétique compatible avec le syndrome d'Apert..
En revanche, lorsque les signes sont subtils ou n'ont pas été identifiés avant la naissance, il est ensuite possible d'effectuer une analyse physique détaillée et divers tests génétiques pour confirmer le diagnostic..
Bien qu'il n'y ait pas de remède spécifique pour le syndrome d'Apert, diverses approches ont été décrites pour traiter les symptômes et les complications médicales de cette pathologie..
Les interventions thérapeutiques les plus efficaces sont celles qui sont mises en œuvre tôt, dans les premiers instants de la vie et impliquent des professionnels de différents domaines.
En règle générale, le traitement des enfants affectés nécessite une planification individualisée, avec plusieurs chirurgies programmées. Ainsi, la prise en charge de cette pathologie repose sur la correction des malformations squelettiques et cranio-faciales, et un accompagnement psychologique et neuropsychologique..
Grâce à la neurochirurgie, l'objectif est de reconstruire la voûte crânienne, tandis que les spécialistes en chirurgie maxillo-faciale tentent de corriger les malformations du visage. Par contre, la participation de chirurgiens traumatologues est également fréquente, pour la reconstruction des malformations présentes dans les mains et les pieds.
De plus, la conception de programmes individualisés de stimulation précoce, de réadaptation de la communication, de formation aux compétences sociales ou de suivi psychopédagogique, sont bénéfiques pour la réalisation d'un développement optimal, fonctionnel et indépendant des personnes touchées..


Personne n'a encore commenté ce post.